本系列持续更新Seurat单细胞分析教程,欢迎关注!
非线形降维
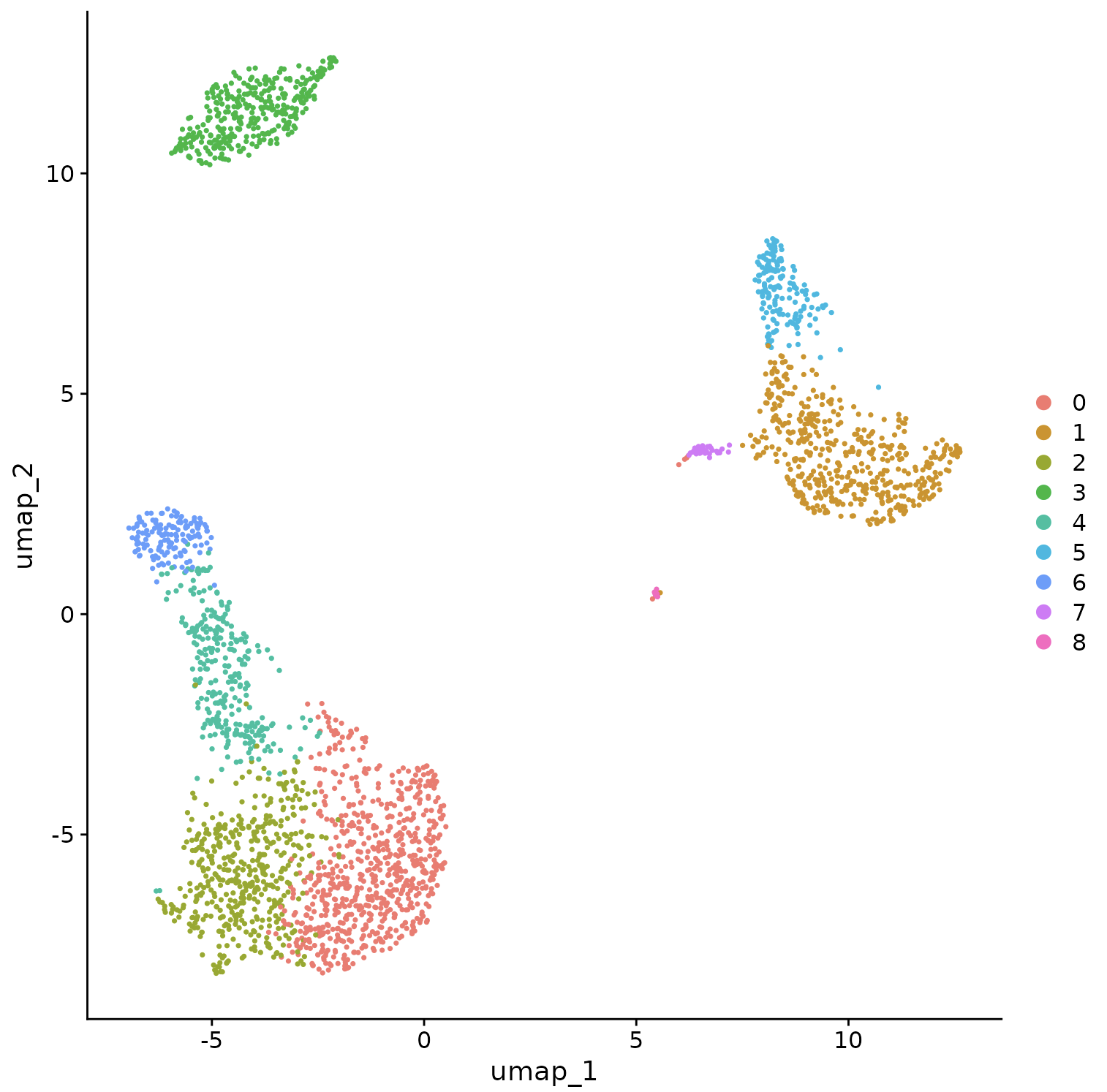
Seurat 提供了几种非线性降维技术,例如 tSNE 和 UMAP,来可视化和探索这些数据集。这些算法的目标是学习数据集中的底层结构,以便将相似的细胞放在低维空间中。因此,在上面确定的基于图的簇内分组在一起的细胞应该在这些降维图上共同定位。
虽然和其他人经常发现 tSNE 和 UMAP 等 2D 可视化技术是探索数据集的有价值的工具,但所有可视化技术都有局限性,并且不能完全代表基础数据的复杂性。特别是,这些方法旨在保留数据集中的局部距离(即确保具有非常相似的基因表达谱的细胞共定位),但通常不会保留更多的全局关系。我们鼓励用户利用 UMAP 等技术进行可视化,但避免仅根据可视化技术得出生物学结论。
pbmc <- RunUMAP(pbmc, dims = 1:10)
# note that you can set `label = TRUE` or use the LabelClusters function to help label
# individual clusters
DimPlot(pbmc, reduction = "umap")
您可以在此时保存对象,以便可以轻松地重新加载它,而无需重新运行上面执行的计算密集型步骤,或者轻松地与协作者共享。
寻找差异表达特征(簇生物标志物)
Seurat 可以帮助您找到通过差异表达 (DE) 定义簇的标记。默认情况下,与所有其他细胞相比,它识别单个簇的阳性和阴性标记(在 ident.1 中指定)。 FindAllMarkers() 会针对所有集群自动执行此过程,但您也可以测试集群组之间的对比,或针对所有细胞进行测试。
在 Seurat v5 中,我们使用 presto 软件包来显着提高 DE 分析的速度,特别是对于大型数据集。对于不使用 presto 的用户,您可以查看该函数的文档(?FindMarkers)来探索 min.pct 和 logfc.threshold 参数,可以增加这些参数以提高 DE 测试的速度。
# find all markers of cluster 2
cluster2.markers <- FindMarkers(pbmc, ident.1 = 2)
head(cluster2.markers, n = 5)
## p_val avg_log2FC pct.1 pct.2 p_val_adj
## IL32 2.593535e-91 1.3221171 0.949 0.466 3.556774e-87
## LTB 7.994465e-87 1.3450377 0.981 0.644 1.096361e-82
## CD3D 3.922451e-70 1.0562099 0.922 0.433 5.379250e-66
## IL7R 1.130870e-66 1.4256944 0.748 0.327 1.550876e-62
## LDHB 4.082189e-65 0.9765875 0.953 0.614 5.598314e-61
# find all markers distinguishing cluster 5 from clusters 0 and 3
cluster5.markers <- FindMarkers(pbmc, ident.1 = 5, ident.2 = c(0, 3))
head(cluster5.markers, n = 5)
## p_val avg_log2FC pct.1 pct.2 p_val_adj
## FCGR3A 2.150929e-209 6.832372 0.975 0.039 2.949784e-205
## IFITM3 6.103366e-199 6.181000 0.975 0.048 8.370156e-195
## CFD 8.891428e-198 6.052575 0.938 0.037 1.219370e-193
## CD68 2.374425e-194 5.493138 0.926 0.035 3.256286e-190
## RP11-290F20.3 9.308287e-191 6.335402 0.840 0.016 1.276538e-186
# find markers for every cluster compared to all remaining cells, report only the positive
# ones
pbmc.markers <- FindAllMarkers(pbmc, only.pos = TRUE)
pbmc.markers %>%
group_by(cluster) %>%
dplyr::filter(avg_log2FC > 1)
## # A tibble: 7,046 × 7
## # Groups: cluster [9]
## p_val avg_log2FC pct.1 pct.2 p_val_adj cluster gene
## <dbl> <dbl> <dbl> <dbl> <dbl> <fct> <chr>
## 1 1.74e-109 1.19 0.897 0.593 2.39e-105 0 LDHB
## 2 1.17e- 83 2.37 0.435 0.108 1.60e- 79 0 CCR7
## 3 8.94e- 79 1.09 0.838 0.403 1.23e- 74 0 CD3D
## 4 3.05e- 53 1.02 0.722 0.399 4.19e- 49 0 CD3E
## 5 3.28e- 49 2.10 0.333 0.103 4.50e- 45 0 LEF1
## 6 6.66e- 49 1.25 0.623 0.358 9.13e- 45 0 NOSIP
## 7 9.31e- 44 2.02 0.328 0.11 1.28e- 39 0 PRKCQ-AS1
## 8 4.69e- 43 1.53 0.435 0.184 6.43e- 39 0 PIK3IP1
## 9 1.47e- 39 2.70 0.195 0.04 2.01e- 35 0 FHIT
## 10 2.44e- 33 1.94 0.262 0.087 3.34e- 29 0 MAL
## # ℹ 7,036 more rows
Seurat 有几种差异表达测试,可以使用 test.use 参数进行设置。例如,ROC 测试返回任何单个标记的“分类能力”(范围从 0 到 1)。
cluster0.markers <- FindMarkers(pbmc, ident.1 = 0, logfc.threshold = 0.25, test.use = "roc", only.pos = TRUE)
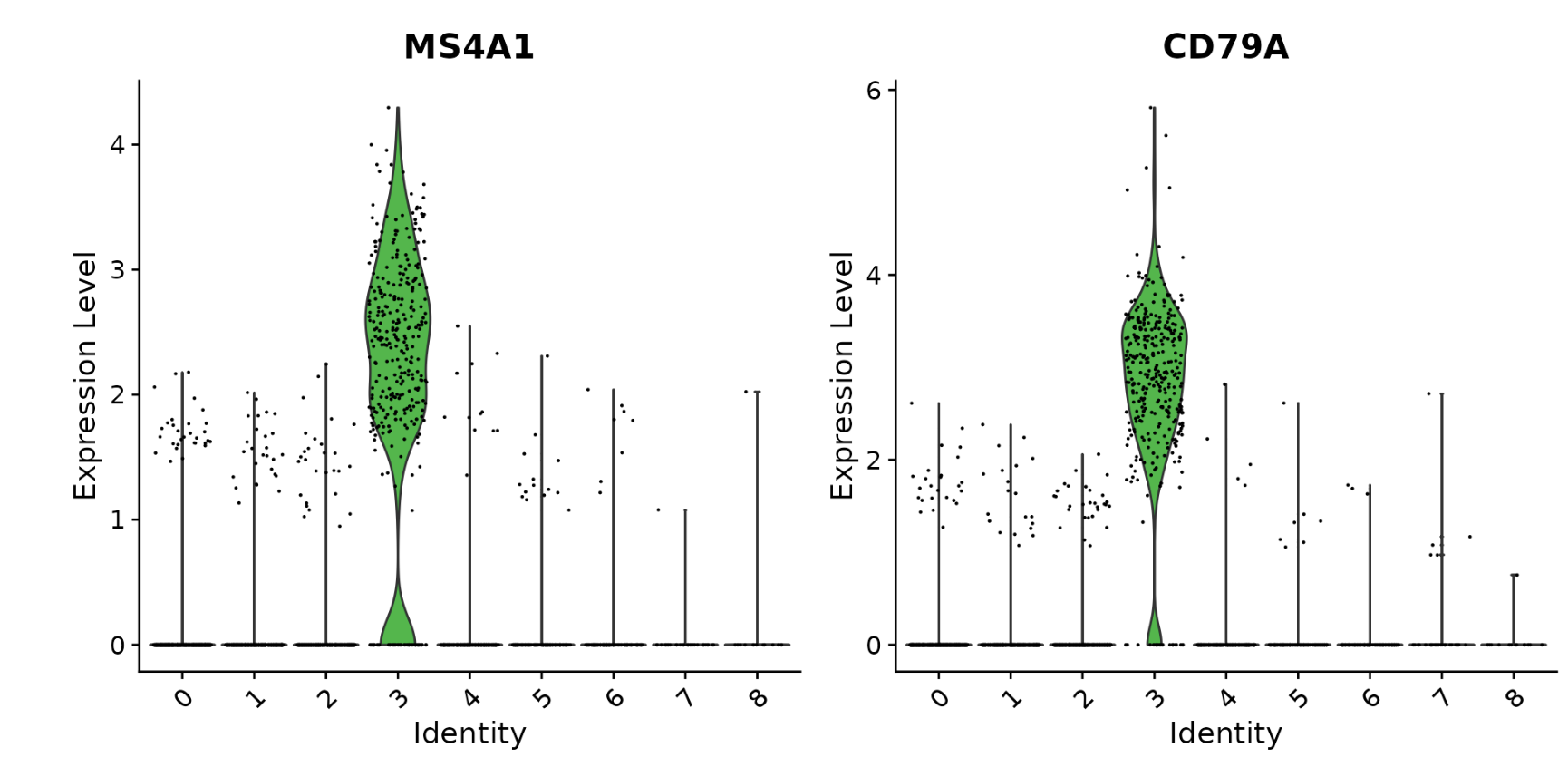
我们提供了几种用于可视化标记表达的工具。 VlnPlot()(显示跨簇的表达概率分布)和 FeaturePlot()(在 tSNE 或 PCA 图上可视化特征表达)是我们最常用的可视化。我们还建议探索 RidgePlot()、CellScatter() 和 DotPlot() 作为查看数据集的附加方法。
VlnPlot(pbmc, features = c("MS4A1", "CD79A"))
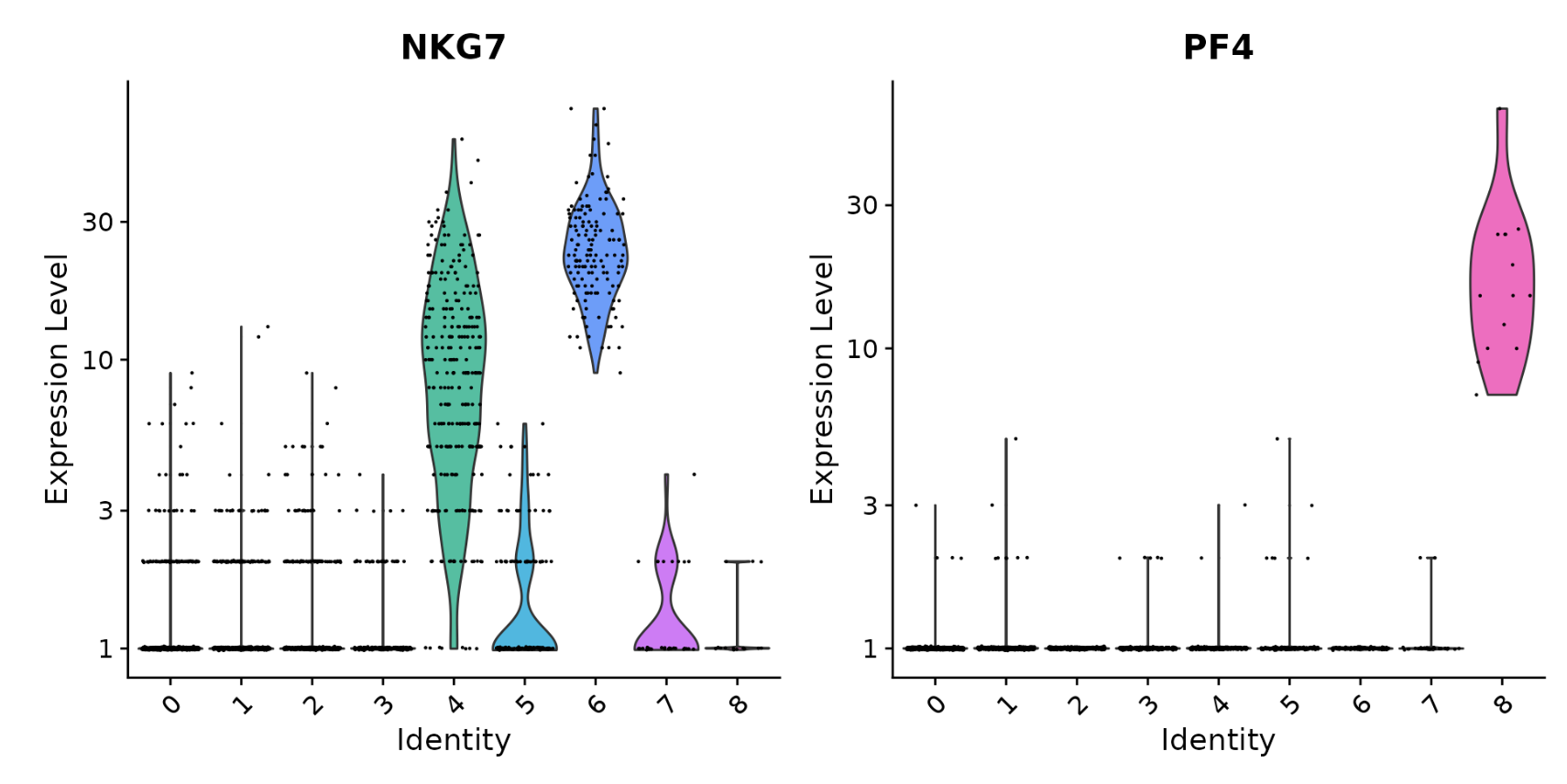
# you can plot raw counts as well
VlnPlot(pbmc, features = c("NKG7", "PF4"), slot = "counts", log = TRUE)
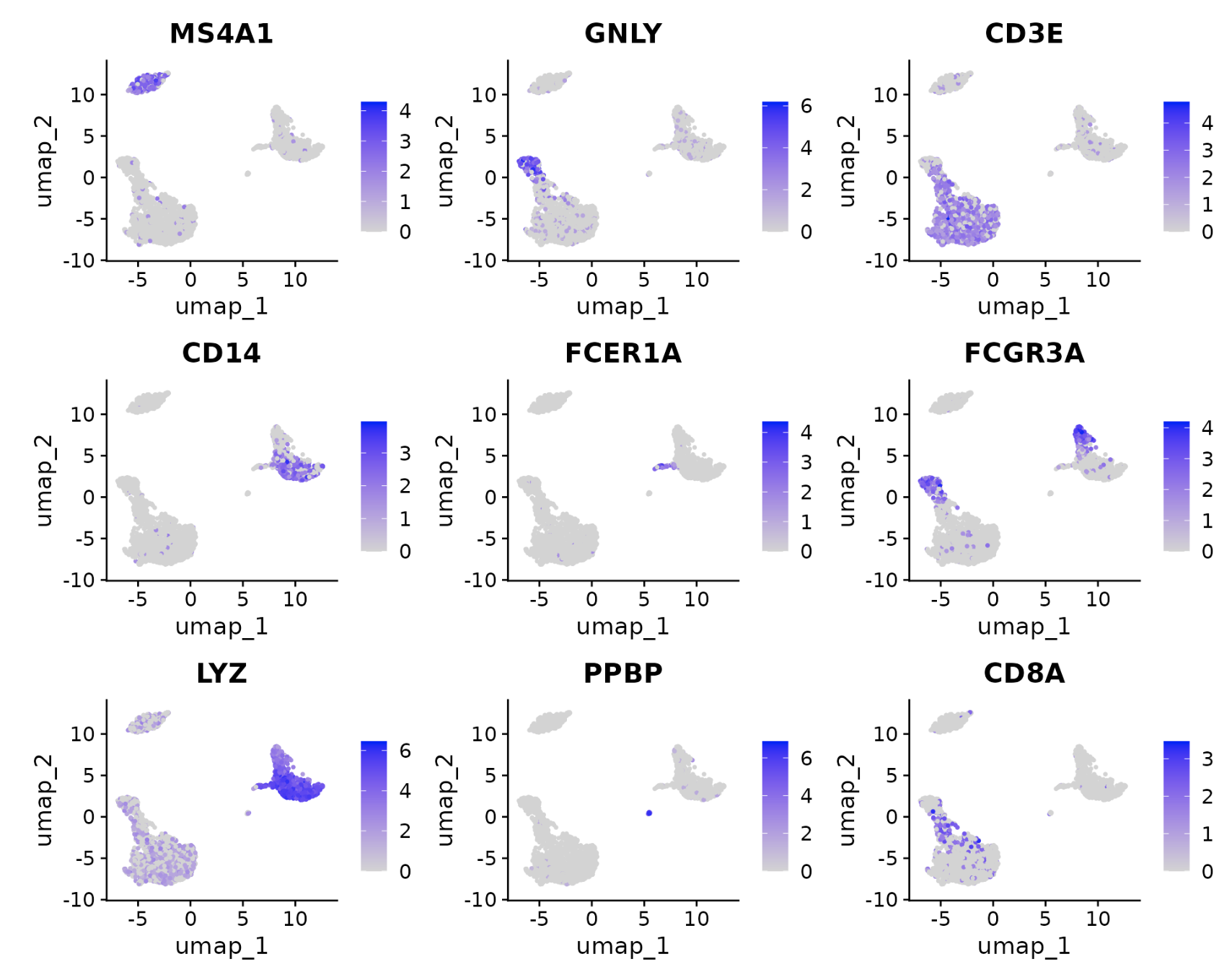
FeaturePlot(pbmc, features = c("MS4A1", "GNLY", "CD3E", "CD14", "FCER1A", "FCGR3A", "LYZ", "PPBP",
"CD8A"))
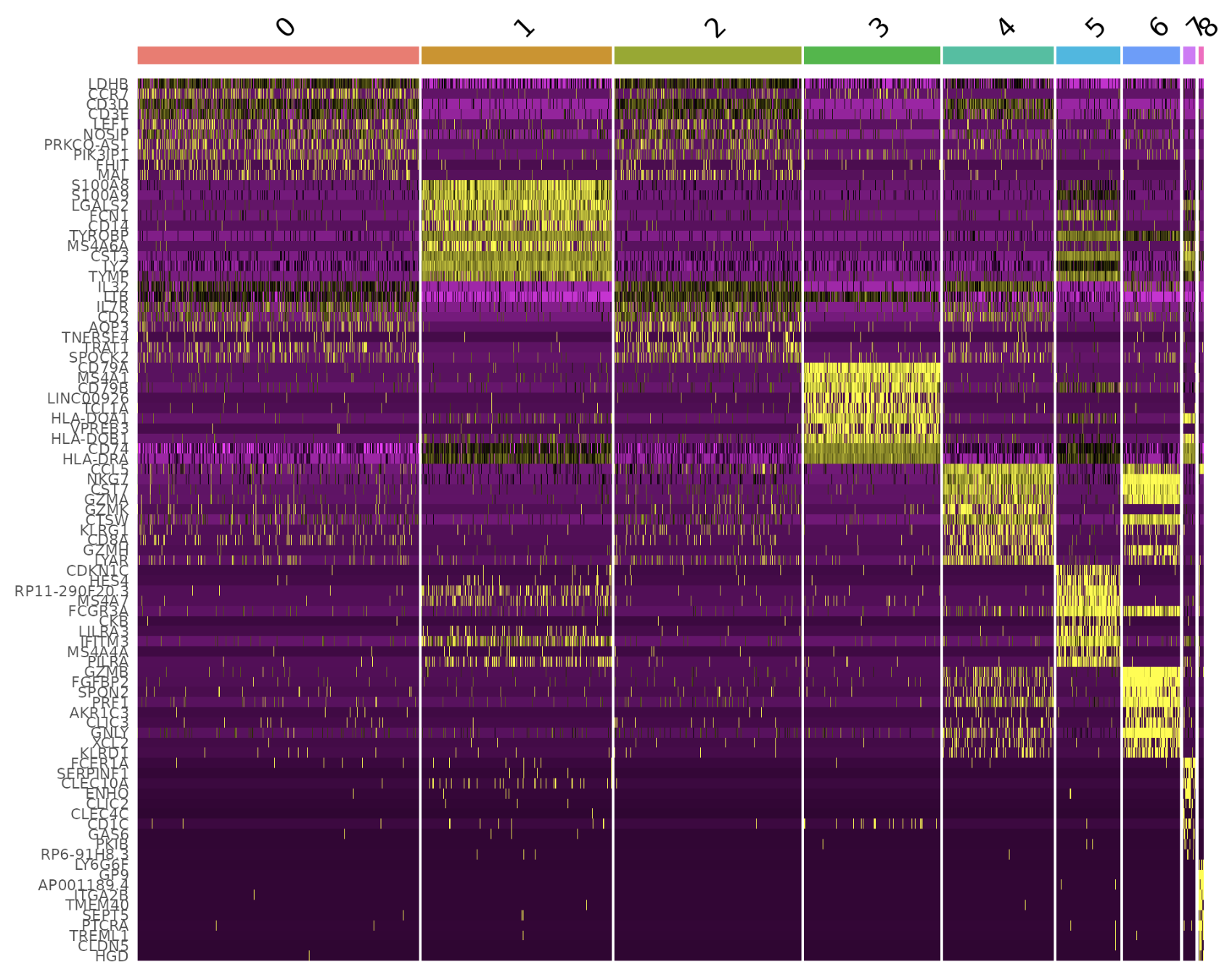
DoHeatmap() 为给定的细胞和特征生成表达式热图。在本例中,我们绘制每个簇的前 20 个标记(如果少于 20 个则为所有标记)。
pbmc.markers %>%
group_by(cluster) %>%
dplyr::filter(avg_log2FC > 1) %>%
slice_head(n = 10) %>%
ungroup() -> top10
DoHeatmap(pbmc, features = top10$gene) + NoLegend()
细胞类型分配
在此数据集的情况下,可以使用规范标记轻松地将无偏聚类与已知细胞类型进行匹配:
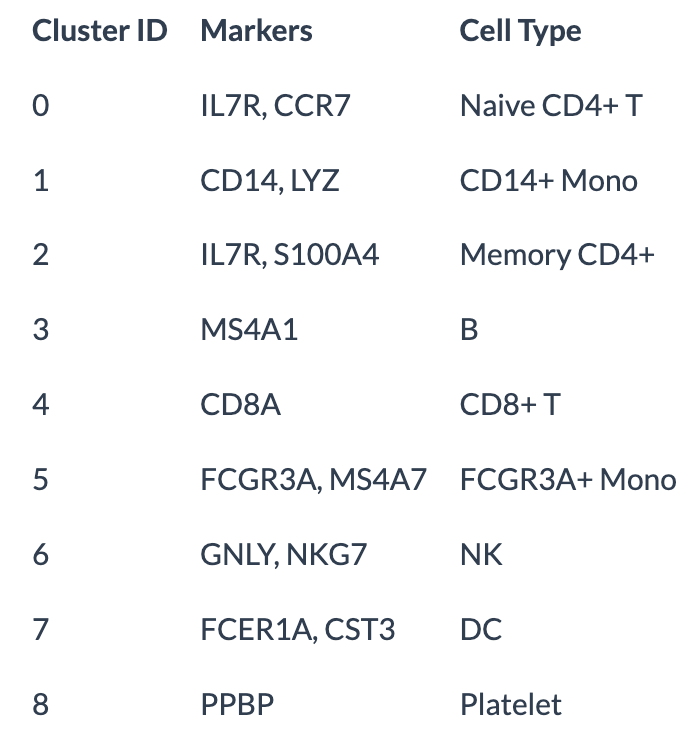
new.cluster.ids <- c("Naive CD4 T", "CD14+ Mono", "Memory CD4 T", "B", "CD8 T", "FCGR3A+ Mono",
"NK", "DC", "Platelet")
names(new.cluster.ids) <- levels(pbmc)
pbmc <- RenameIdents(pbmc, new.cluster.ids)
DimPlot(pbmc, reduction = "umap", label = TRUE, pt.size = 0.5) + NoLegend()
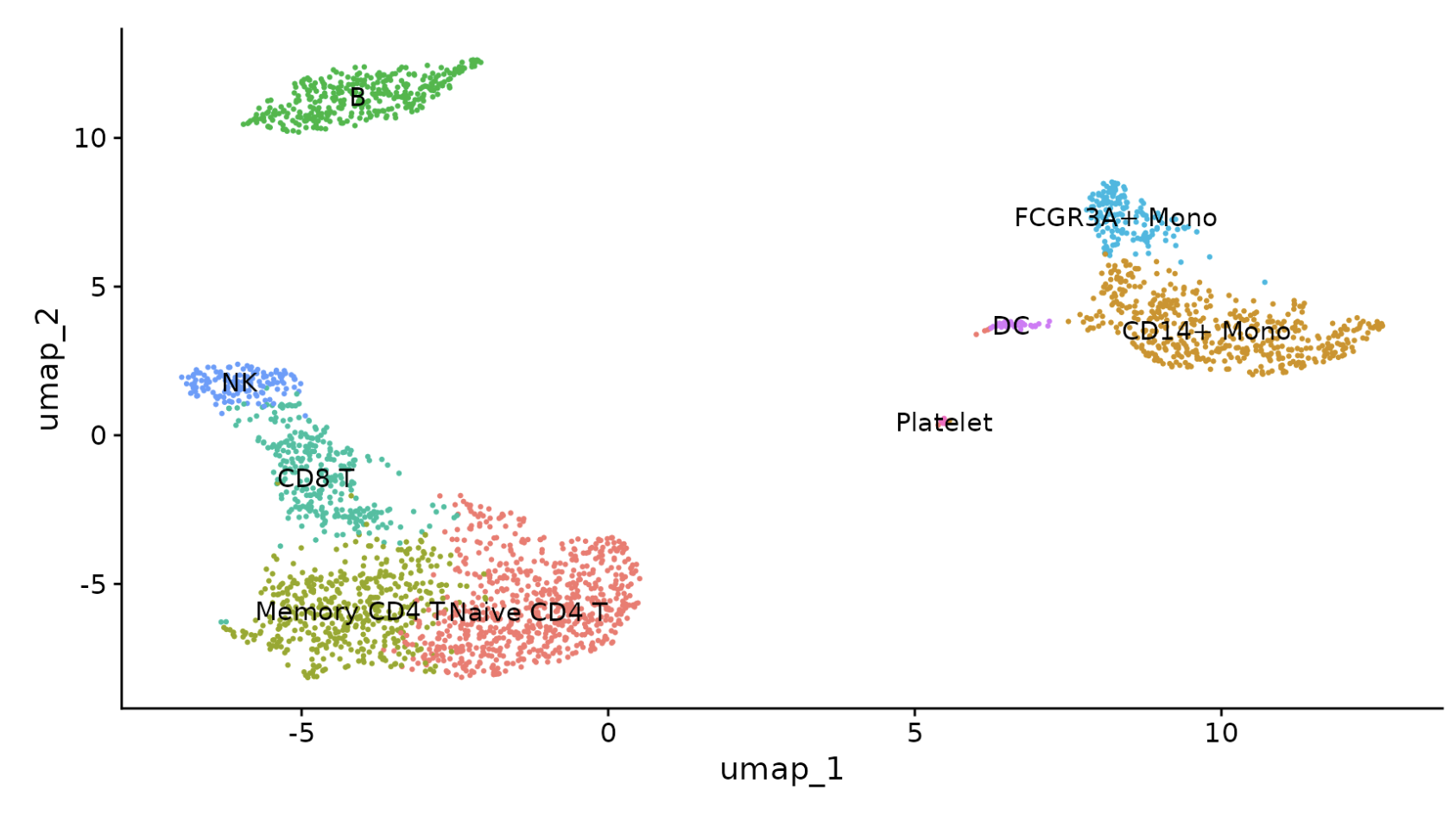
library(ggplot2)
plot <- DimPlot(pbmc, reduction = "umap", label = TRUE, label.size = 4.5) + xlab("UMAP 1") + ylab("UMAP 2") +
theme(axis.title = element_text(size = 18), legend.text = element_text(size = 18)) + guides(colour = guide_legend(override.aes = list(size = 10)))
ggsave(filename = "../output/images/pbmc3k_umap.jpg", height = 7, width = 12, plot = plot, quality = 50)
saveRDS(pbmc, file = "../output/pbmc3k_final.rds")
未完待续,持续更新,欢迎关注!
本文由 mdnice 多平台发布

)

机器人栅格路径规划,输出做短路径图和适应度曲线。)

)



)





)
)

)
