数据质控
将SRR转为fastq之后,我们需要对fastq进行质量检查,排除质量不好的数据
1.质量检查,生成报告文件
ls *fastq.gz|while read id;do fastqc $id;done
并行处理
ls *fastq.gz|xargs fastqc -t 10
2.生成 html 报告文件和对应的 zip 压缩文件,并通过 scp 命令传输到本地后用浏览器打开查看。
#传文件
scp -i username@server-ip:~/my_project/airway/QC_results /Users/yangshengyu/qc#传文件夹
scp -r username@server-ip:~/my_project/airway/QC_results /Users/yangshengyu/qc
#如果默认端口22关闭,使用-P指定端口多个报告文件合成一个总的报告文件方便查看,不用一个个打开检查
mkdir QC_results
mv *zip *html QC_results
cd QC_results
multiqc ./
3.结果说明
FastQC 结果由11个模块组成,对于结果报告各个模块的说明参考FastQC 文档
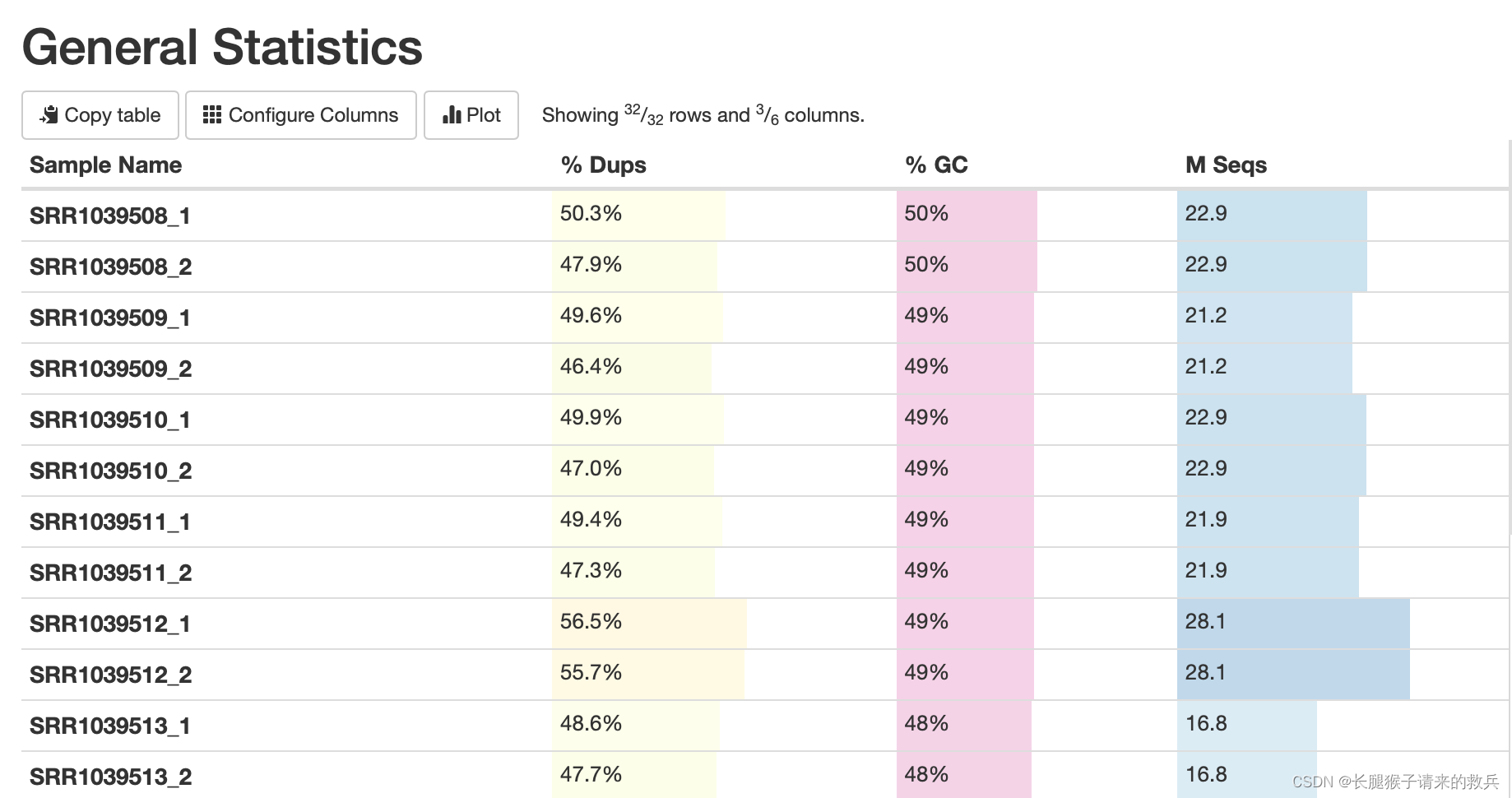
1)综合统计(General Statistics)
重复reads的比例(%Dups)、GC含量占总碱基的比例、总测序量(M Seqs,单位:millions)
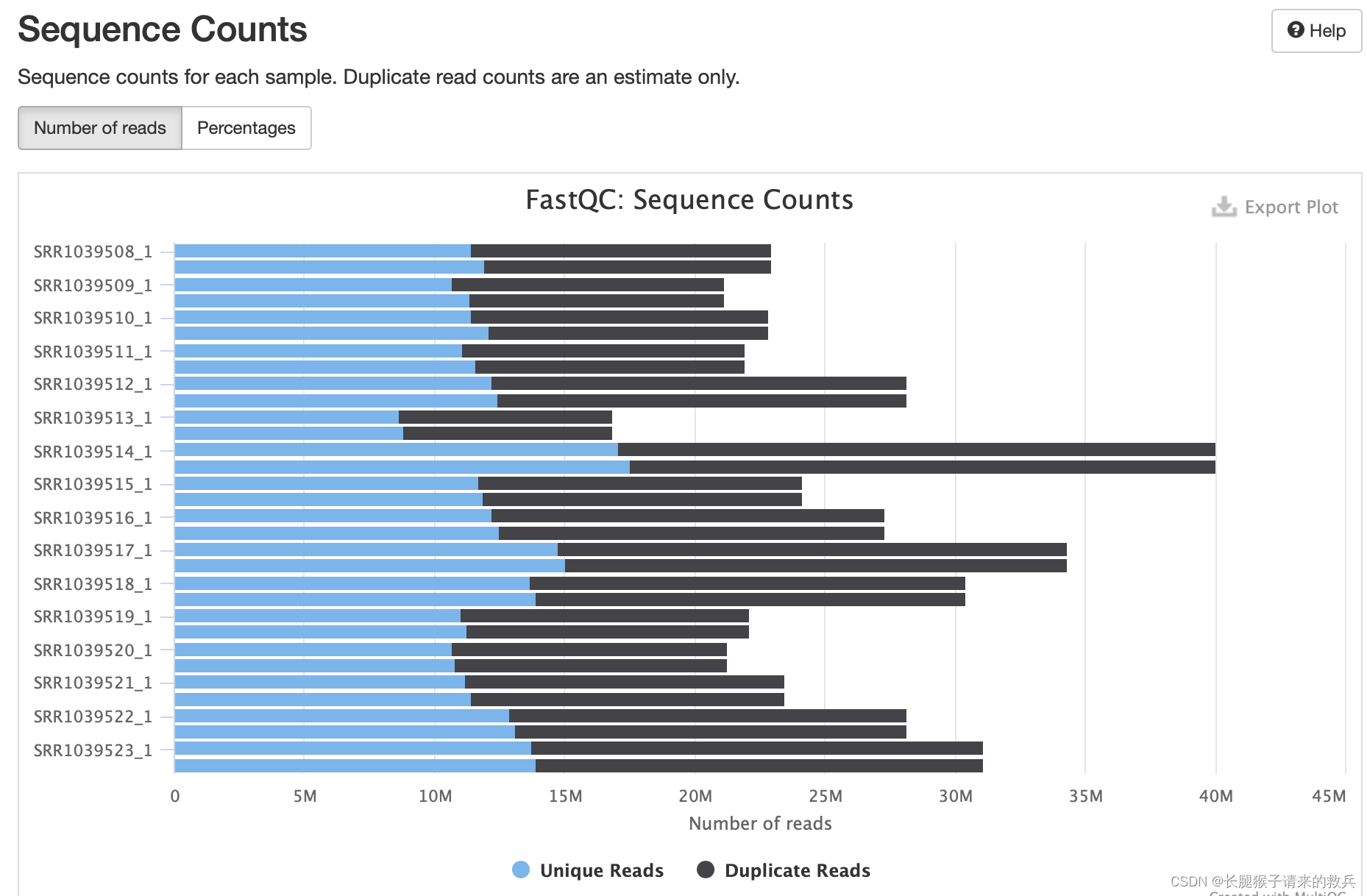
2)序列的计数(sequence counts)
可以看到reads的数量和重复reads的百分比
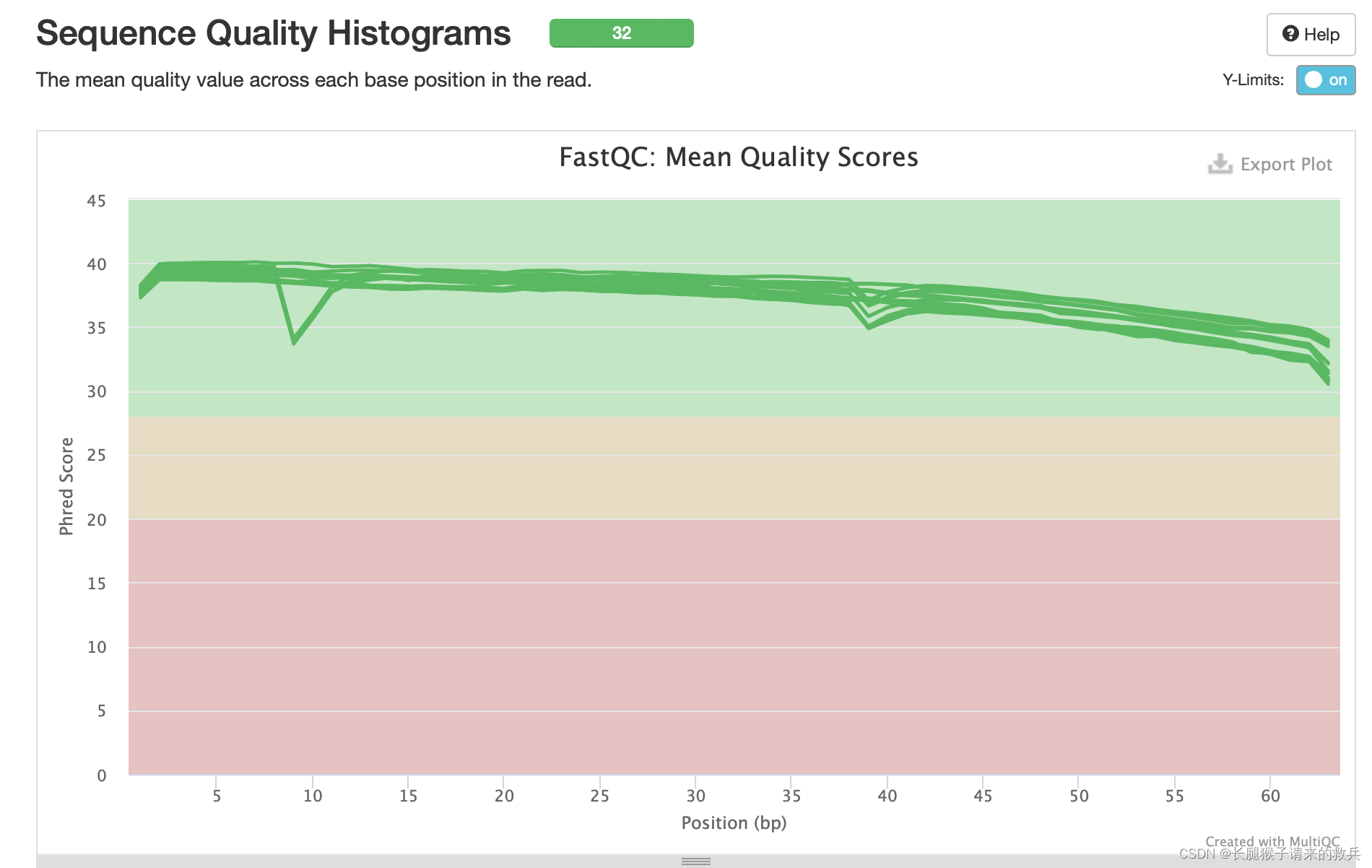
3)每个read各位置碱基的平均测序质量
横坐标——碱基的位置
纵坐标——质量分数=-10log10p(p代表错误率),所以当质量分数为40的时候,p就是0.0001。此时说明测序质量非常好。
绿色区间——质量很好,橙色区间——质量合理,红色区间——质量不好。
由此可知,32个样本在60个碱基前的测序质量平均线都在绿色区域内,质量很好。
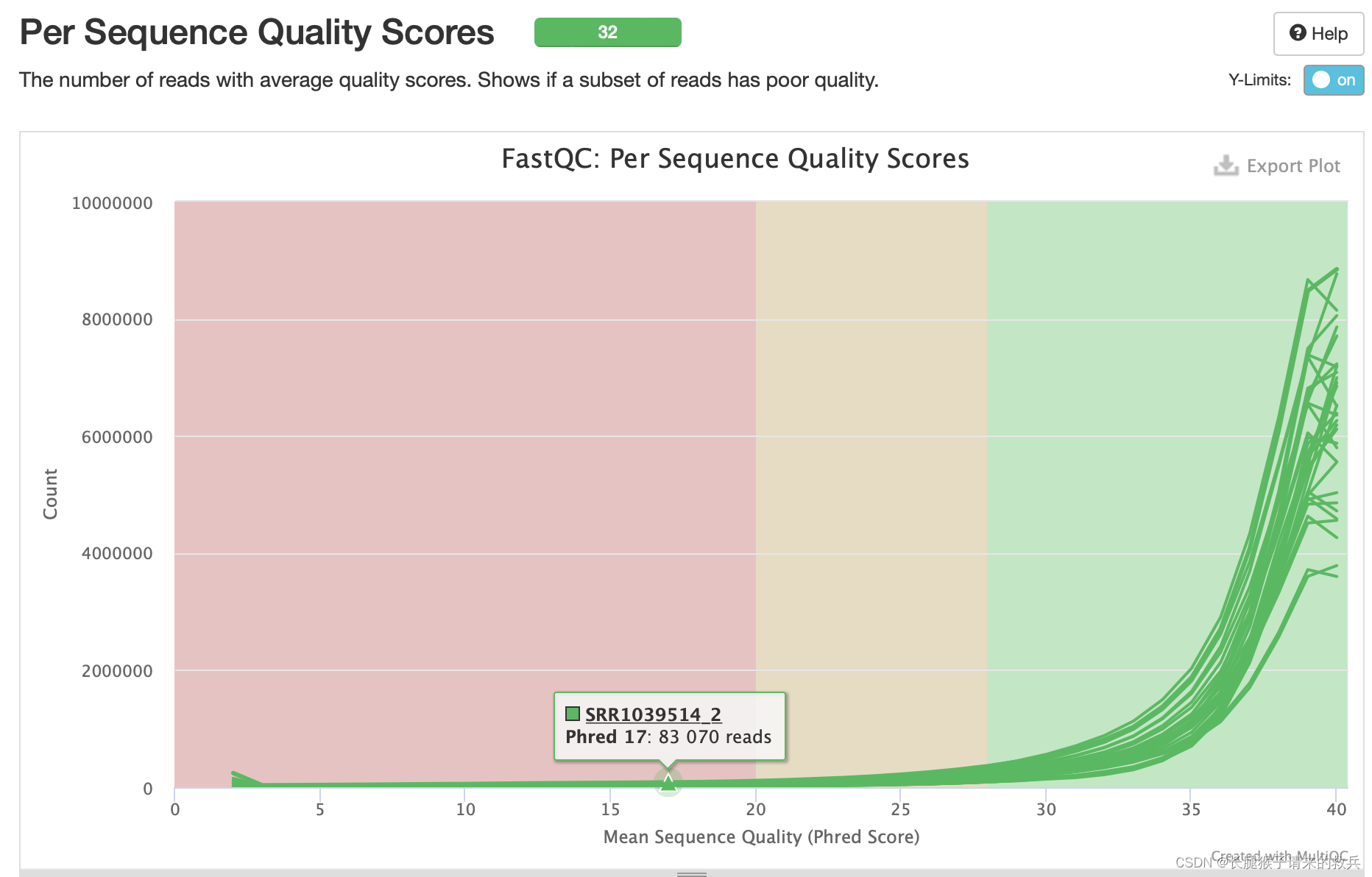
4)具有平均质量分数的reads的数量
绿色区间——质量很好,橙色区间——质量合理,红色区间——质量不好。由此可知,32个样本大部分都在绿色区域内,质量很好。
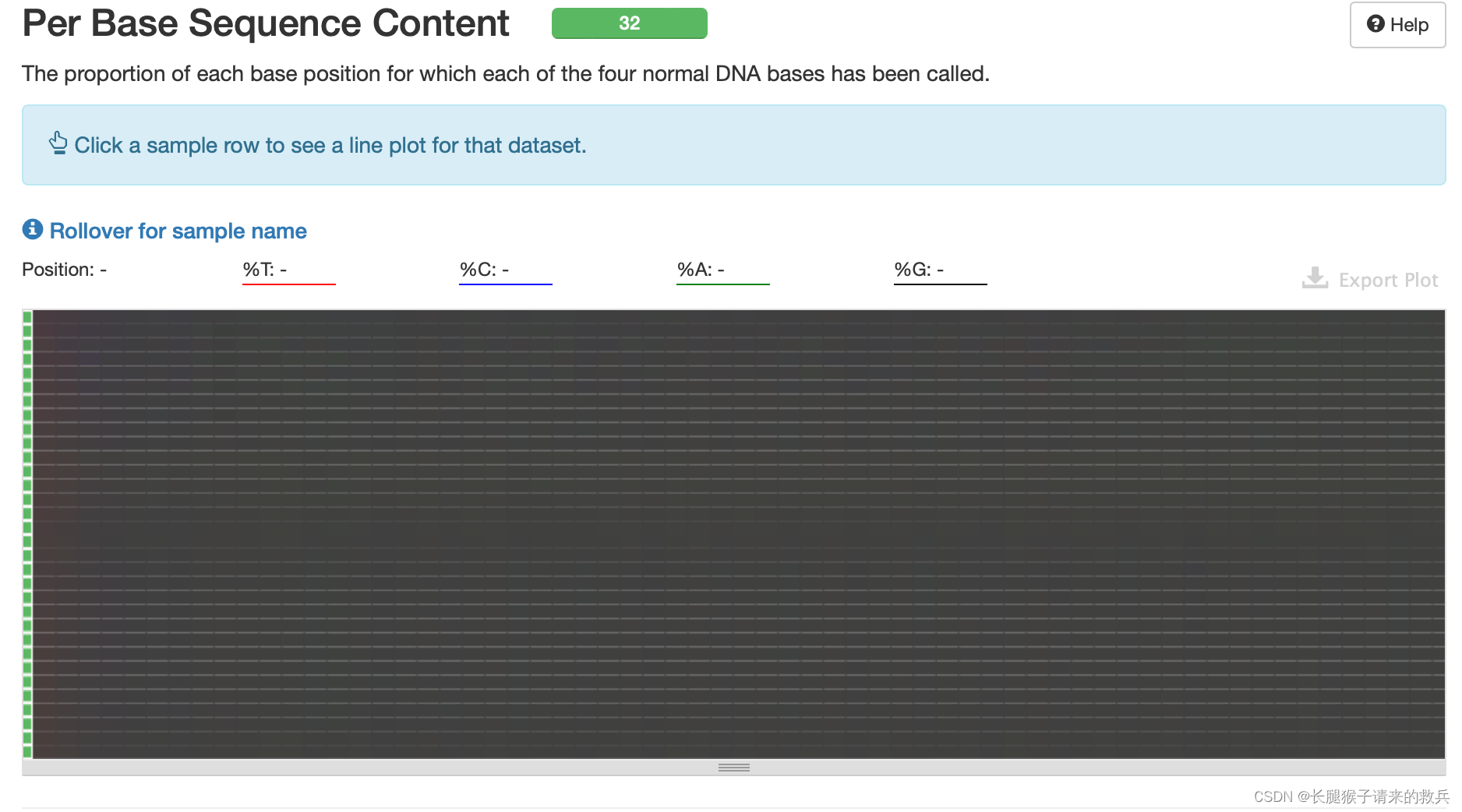
5)每个read各位置碱基ATCG的比列
reads每个位置的颜色显示由4种颜色的比例混合而成,哪一个碱基的比例大,则趋近于这个碱基所代表的颜色。正常情况下每个位置每种碱基出现的概率是相近的。由下图可知32个样本的ATCG的含量比例是比较均匀的,测序质量是可以的。
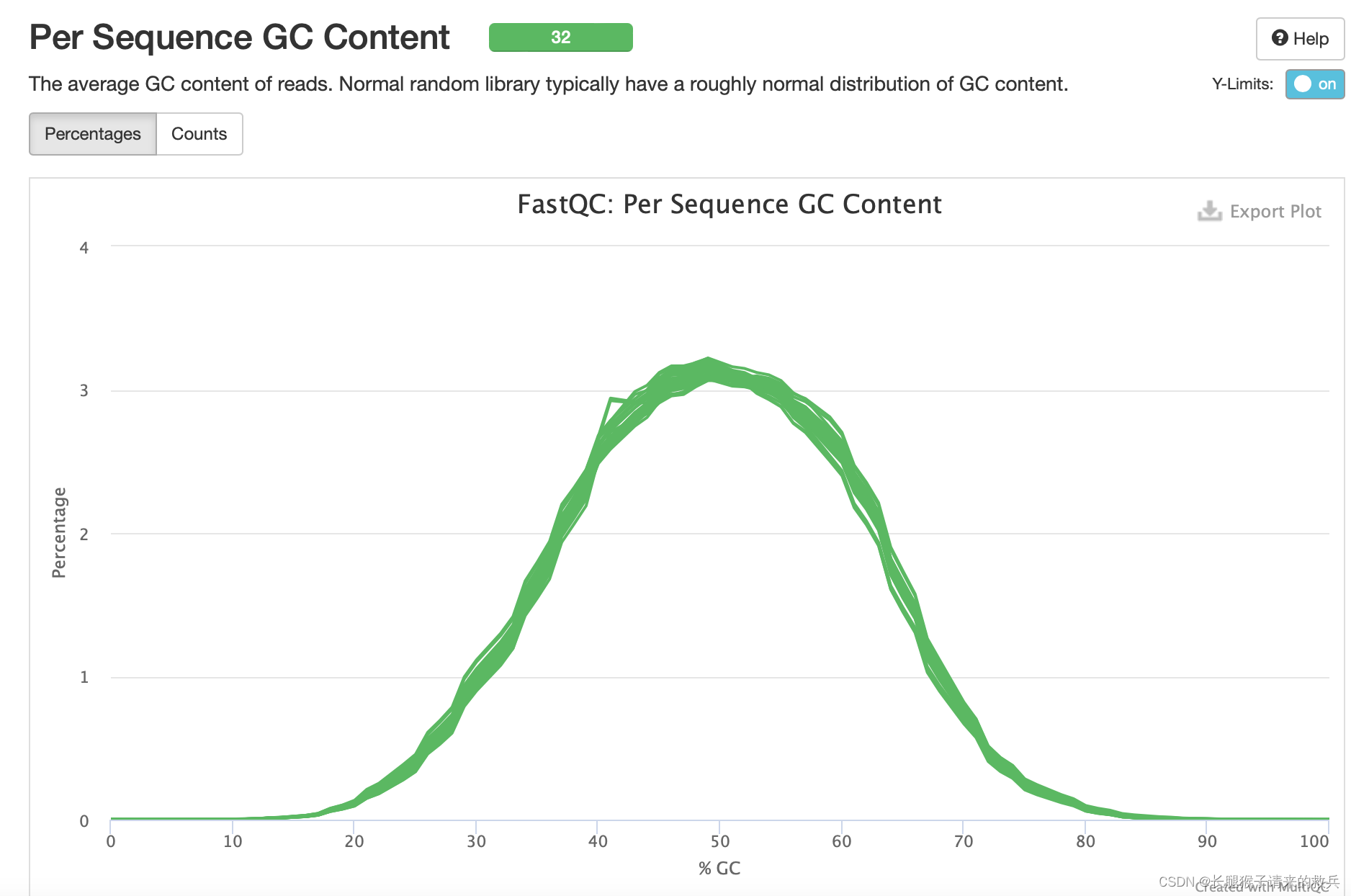
6)reads的平均GC含量
正常的样本的GC含量曲线会趋近于正态分布曲线。由下图可知GC含量曲线符合正态分布曲线,测序质量可以。
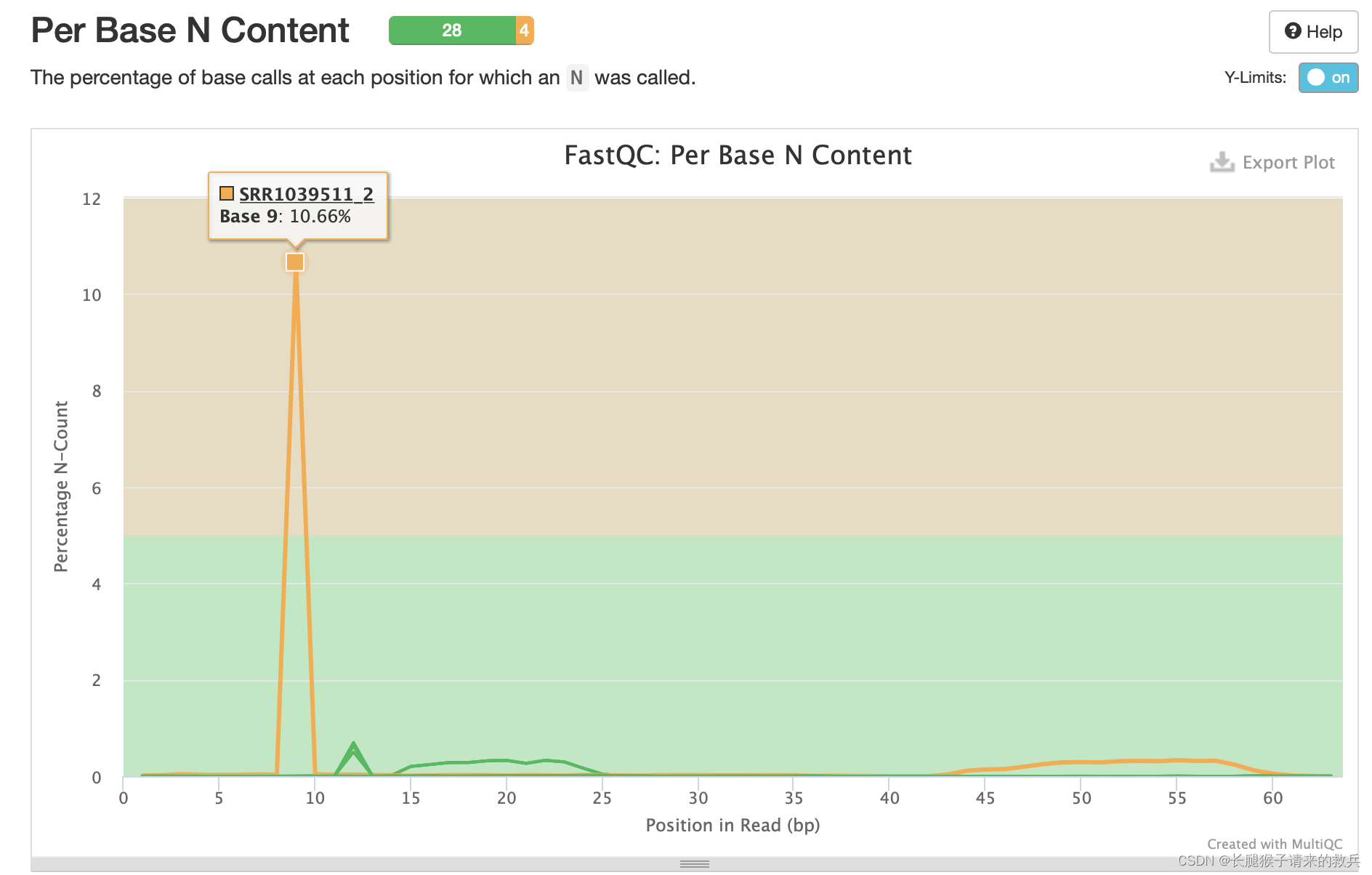
7)每条reads各位置N碱基含量比例
当测序仪器不能辨别某条reads的某个位置到底是什么碱基时,就会产生“N”。正常情况下,N值非常小。由下图可知有样本出现N碱基,其中SRR1039511_2出现的最多。
8)序列长度的分布
所有样本的序列都是单一长度(63bp)

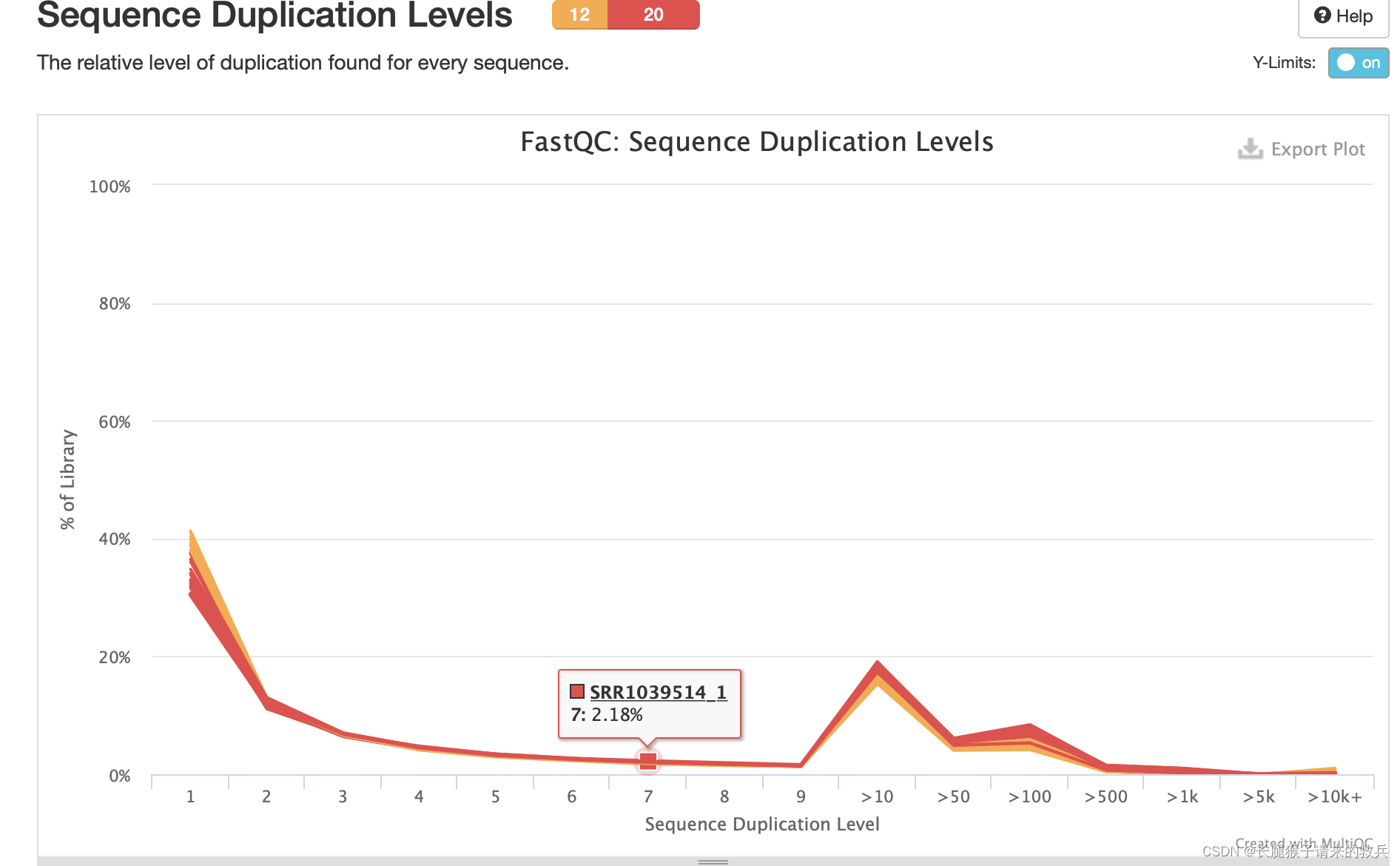
9)每个序列的相对重复水平
横坐标:每个序列的相对重复水平
纵坐标:在文库中的比例
由下图可知每个样本序列的相对重复水平都较高,测序质量不好。
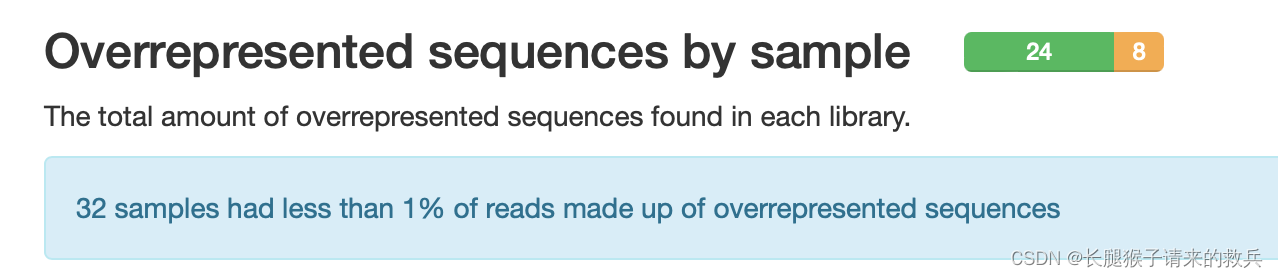
10)文库中过表达序列的比例
横坐标——过表达序列的比例
一条序列的重复数,因为一个转录组中有非常多的转录本,一条序列再怎么多也不太会占整个转录组的一小部分(比如1%),如果出现这种情况,不是这种转录本巨量表达,就是样品被污染。
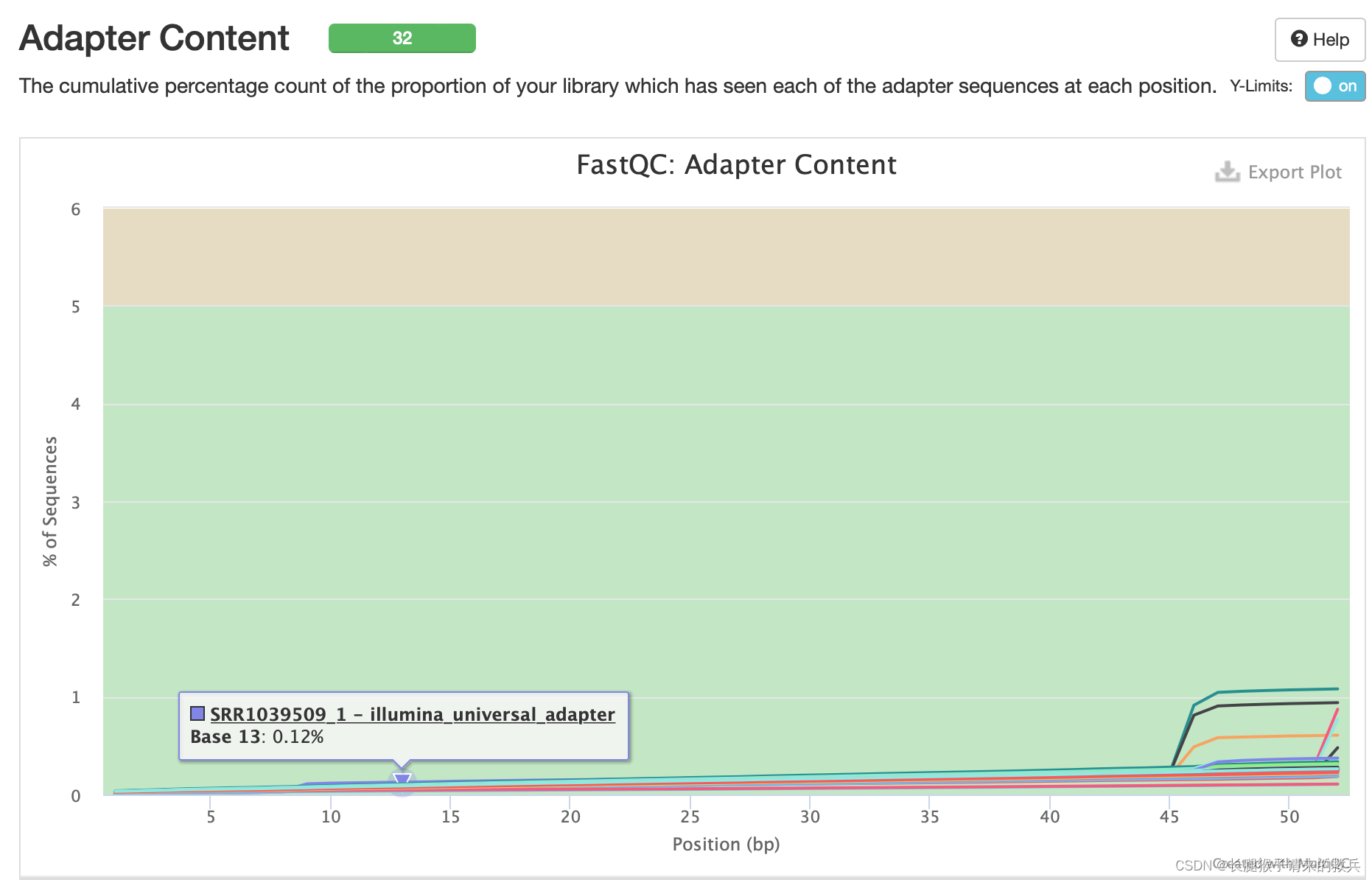
11)接头含量
32个样本的接头含量基本都低于1%
4.原始数据修剪
使用trim_galore对原始数据进行去接头和质控
nohup trim_galore -q 25 --phred33 --length 35 --stringency 3 --fastqc -o ../clean $fq &##批量处理
for fq in `ls |grep fastq$`; do nohup trim_galore -q 25 --phred33 --length 35 --stringency 3 --fastqc -o ../clean $fq ; done &
参数说明:
-q 25 # 设定Phred quality score阈值是25
-phred33 # 指定使用phred33碱基质量值体系
–length 35 # 输出reads长度阈值,小于35bp的reads会被抛弃
–stringency 3 # 可以忍受的前后adapter重叠的碱基数为3
–fastqc # 修剪完数据之后运行fastqc
长腿猴子请来的救兵
写于2023年11月21日 上英语课摸鱼写的














 expects parameter 1 to be string, array given)

——寻找字符串中所有字母异位词的子串)


)