欢迎关注我的CSDN:https://spike.blog.csdn.net/
本文地址:https://spike.blog.csdn.net/article/details/135930139
小分子药物生成是一种利用计算方法自动探索化学空间,寻找具有理想生物活性和药物特性的分子结构的过程。从头设计是一种特殊的小分子药物生成方法,不依赖于已知的化合物库,而是完全从零开始构建分子。从头设计的方法可以分为基于原子、基于片段和基于反应的三种类型,根据分子表示的粒度和复杂度进行区分。基于原子的方法是通过逐步添加原子和键来构建分子,基于片段的方法是通过拼接预先定义的化学片段来构建分子,基于反应的方法是通过模拟化学反应来构建分子。从头设计的方法可以结合人工智能和深度学习技术,以提高分子生成的效率和质量。例如,可以使用深度生成模型来学习分子的潜在分布,然后采样或优化生成新的分子。也可以使用强化学习或进化算法来指导分子生成的过程,以满足特定的目标函数。从头设计是药物发现的重要组成部分,可以帮助发现新颖的化学骨架,拓展化学多样性,提高药物创新的潜力。
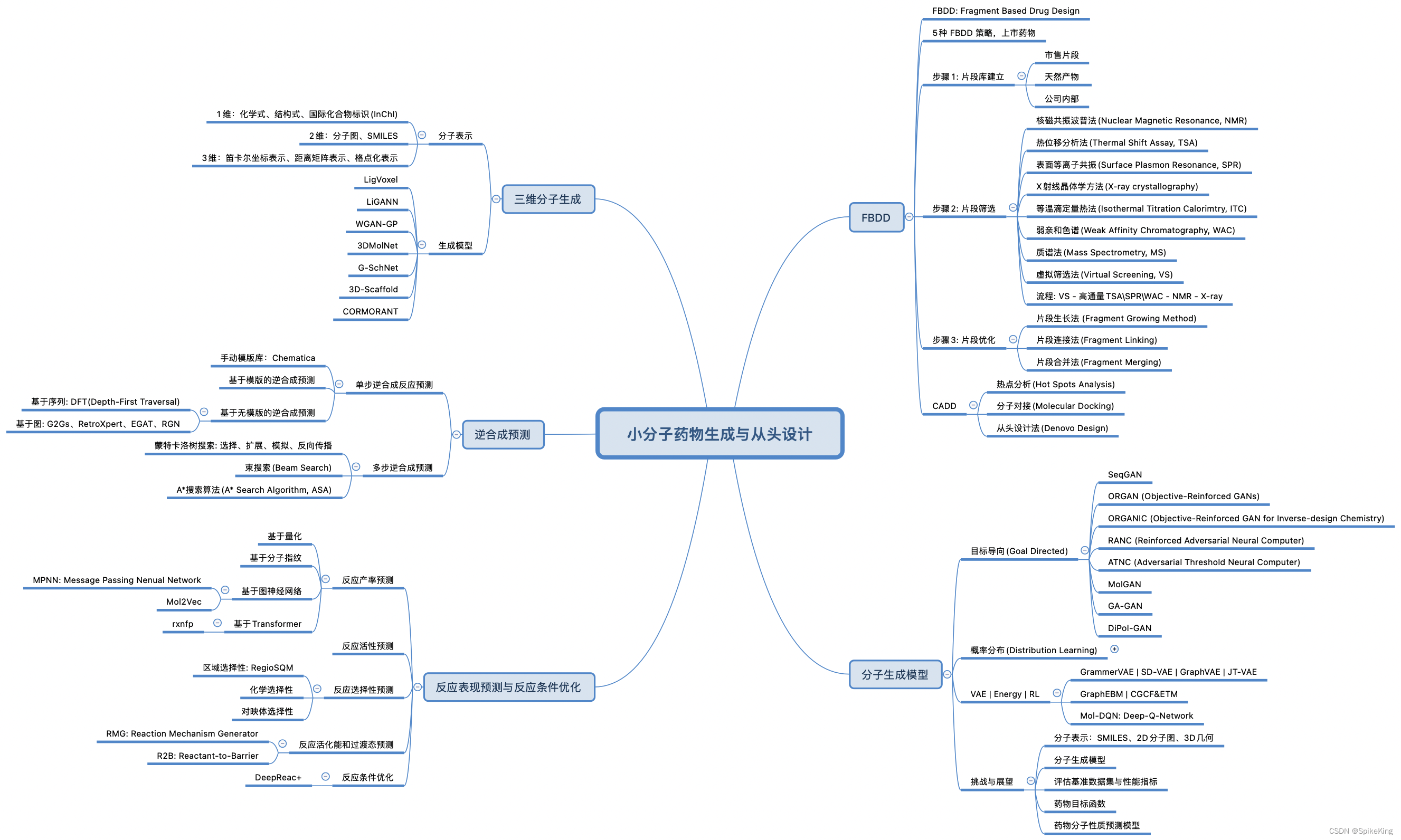
1. 基于片段的药物设计 (FBDD)
基于片段的药物设计(Fragment-Based Drug Design, FBDD)是一种将随机筛选和基于结构的药物设计有机结合的药物发现新方法。基本思想是利用生物物理技术筛选出与靶蛋白有弱相互作用的小分子片段,然后通过片段生长、连接或合并等方法优化这些片段,从而得到具有高亲和力和高选择性的先导化合物。FBDD的优点是可以降低分子淘汰率,提高小分子药物发现效率,拓展化学空间,发现新颖的化学骨架,以及利用配体效率(Ligand Efficiency, LE)等指标评估片段的优化潜力。FBDD的挑战是需要构建合适的片段库,选择有效的筛选方法,以及进行高效的片段优化。FBDD已经成功应用于多个药物靶点和疾病领域,目前已有六种基于FBDD的药物上市,包括治疗癌症的Pexidartinib、Vemurafenib、Erdafitinib、Venetoclax、Sotorasib和Asciminib。
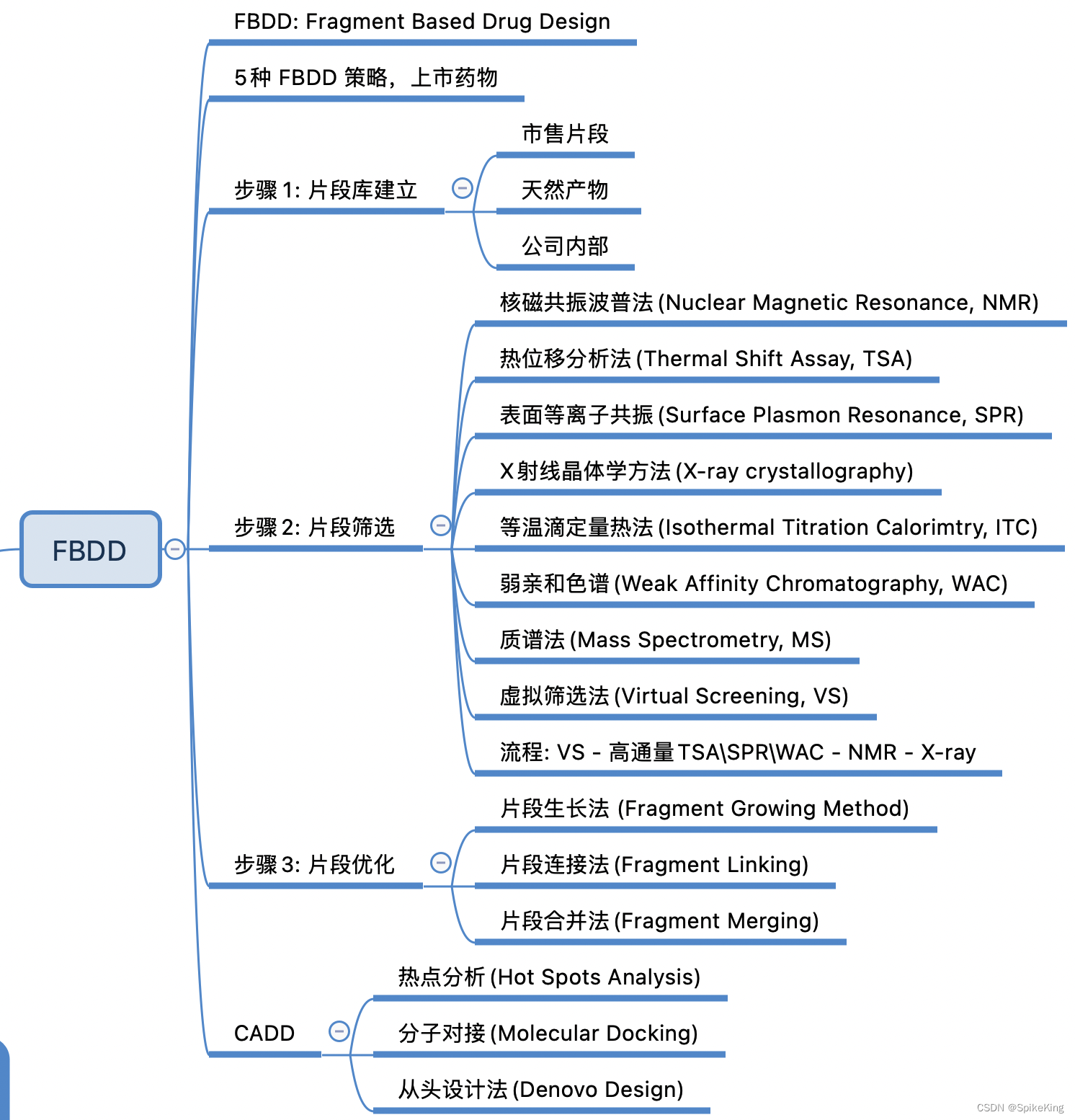
2. 分子生成模型
分子生成模型主要包括3类算法,即 GNN(图神经网络)、VAE(变分自编码器)、RL(强化学习),即:
- GNN(图神经网络)是一种用于处理图结构数据的深度学习模型,可以从分子图中提取分子的拓扑和特征信息,然后用于分子生成或性质预测。GNN的一种常见形式是GCN(图卷积网络),通过对节点和邻居的特征进行加权聚合来更新节点的表示。GNN可以与其他生成模型结合,如VAE、GAN或自回归模型,来实现从头设计或优化分子的目的。
- VAE(变分自编码器)是一种基于潜变量的生成模型,可以将分子映射到一个连续的隐空间,然后从隐空间中采样或优化来生成新的分子。VAE的优点是可以进行隐空间的运算,如插值、重构或属性优化,从而实现分子的探索或改进。VAE的挑战是如何保证生成分子的有效性和多样性,以及如何解决后验分布的坍塌问题。
- RL(强化学习)是一种基于奖励信号的学习方法,可以指导分子生成的过程,以满足特定的目标函数。RL的优点是可以生成具有期望属性的分子,而不需要大量的标注数据。RL的挑战是如何设计合理的奖励函数,以及如何平衡探索和利用的策略。
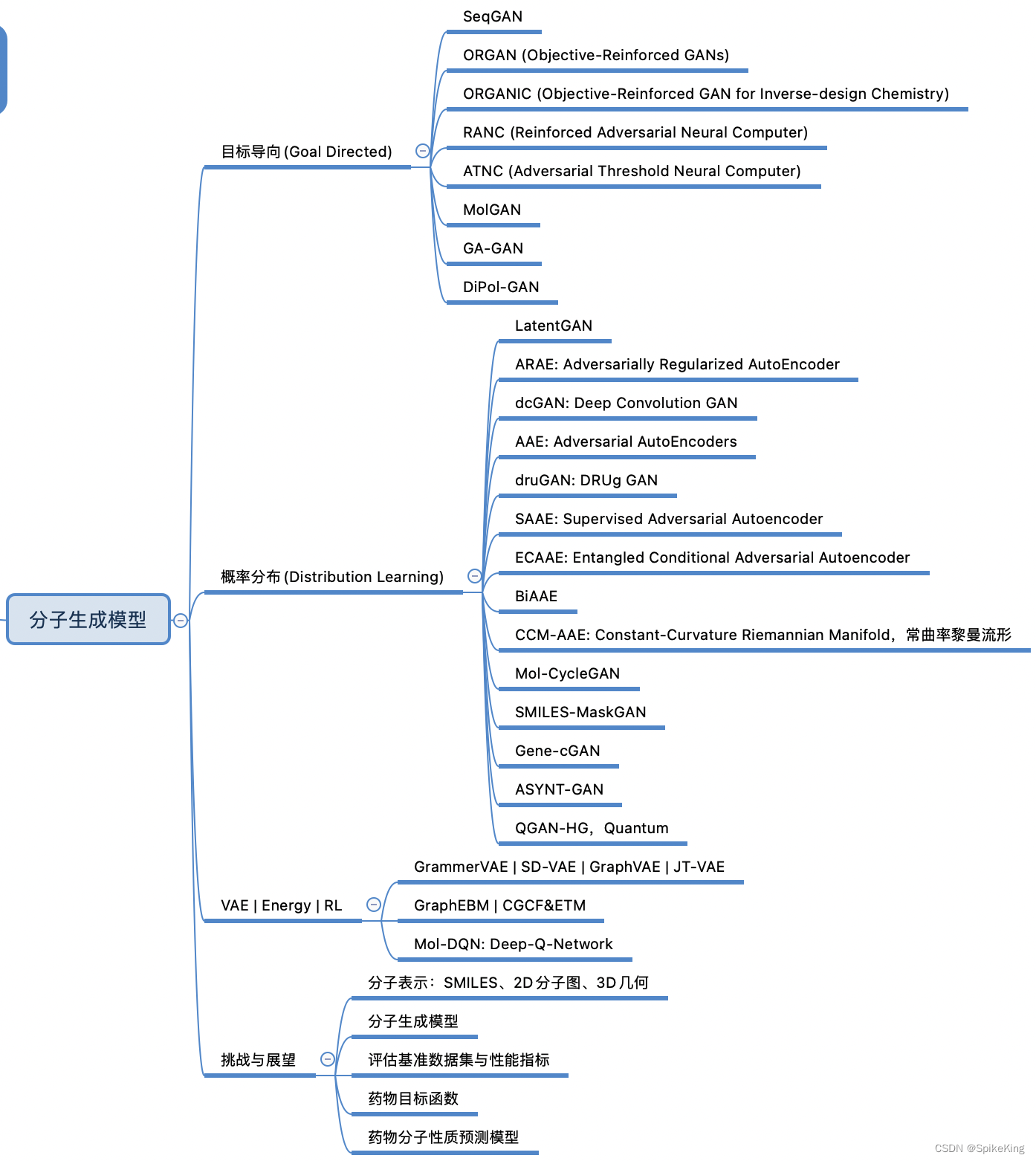
3. 三维分子生成
三维分子生成算法是利用计算方法来自动探索和设计具有三维结构的分子的过程,包括:
- 基于笛卡尔坐标表示的算法:这种算法是通过给定分子中每个原子的三维坐标来描述分子的空间结构,通常使用XYZ文件格式来存储。基于笛卡尔坐标表示的算法可以利用分子力场或量子化学方法来优化分子的几何形状和键角,以及计算分子的性质和活性。基于笛卡尔坐标表示的算法的优点是可以直接反映分子的空间结构,但是需要考虑分子的旋转和平移对坐标的影响。
- 基于距离矩阵表示的算法:这种算法是通过给定分子中每对原子之间的距离来描述分子的空间结构,通常使用二维矩阵来存储。基于距离矩阵表示的算法可以利用距离几何或多维尺度变换等方法来重构分子的三维坐标,以及进行分子的聚类或对齐。基于距离矩阵表示的算法的优点是可以消除分子的旋转和平移的影响,但是不能直接反映分子的几何形状和键角,而且需要满足一定的约束条件,如三角不等式。
- 基于格点化表示的算法:这种算法是通过将分子的空间结构划分为一系列的小立方体(格点)来描述分子的空间结构,通常使用三维张量来存储。基于格点化表示的算法可以利用深度学习或卷积神经网络等方法来学习分子的特征和模式,以及进行分子的生成或优化。基于格点化表示的算法的优点是可以简化分子的空间结构,使其更容易被深度学习模型处理,但是会损失一些分子的细节信息,而且需要确定合适的格点大小和分辨率。
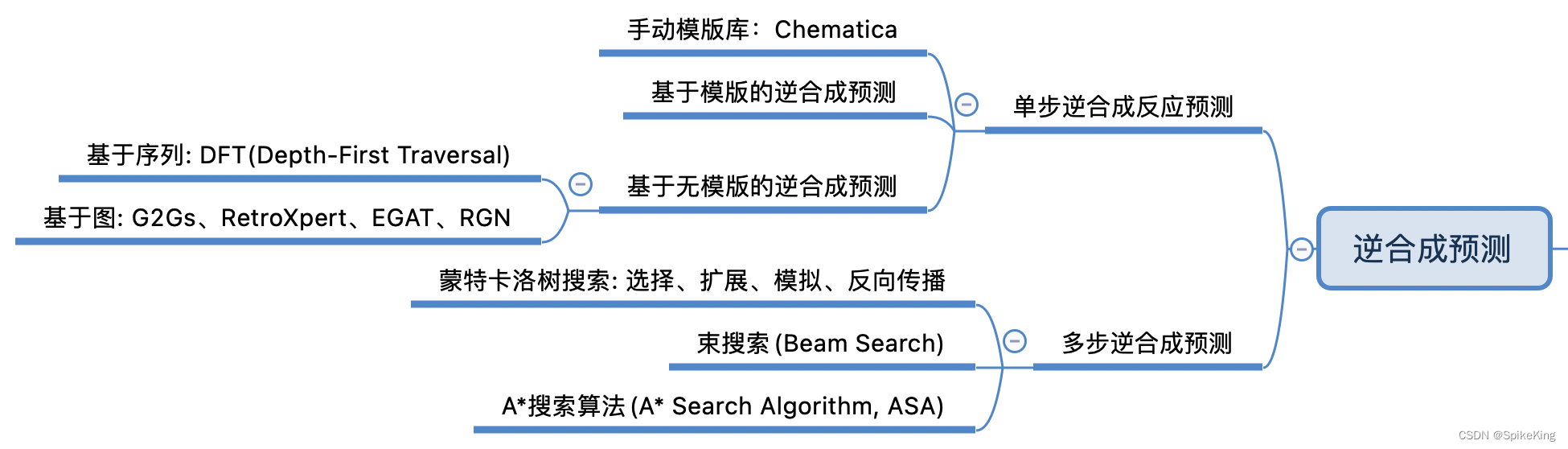
4. 逆合成预测
逆合成预测算法是一种计算机辅助合成规划的方法,可以为给定的目标分子找到一条或多条合成路线,从简单的可购买的前体出发,通过一系列的化学反应,最终得到目标分子。逆合成预测算法可以分为单步逆合成反应预测和多步逆合成预测两种。
- 单步逆合成反应预测是指根据目标分子的结构,预测一种或多种可能的直接前体,以及相应的反应类型和条件。单步逆合成反应预测的方法可以分为基于模板的方法和不依赖模板的方法。基于模板的方法是利用已知的反应规则或模式来匹配目标分子的子结构,从而生成可能的前体。不依赖模板的方法是利用机器学习或深度学习的技术,从大量的反应数据中学习反应的隐含规律和机制,从而生成可能的前体。
- 多步逆合成预测是指根据目标分子的结构,预测一条或多条完整的合成路线,包括每一步的前体、反应类型和条件。多步逆合成预测的方法通常是基于单步逆合成反应预测的方法,结合一种或多种搜索策略,如启发式搜索、蒙特卡罗树搜索、强化学习等,来递归地应用单步预测,直到达到终止条件,如前体的复杂度、可获得性、成本等。
逆合成预测算法的应用可以帮助化学家设计更有效、更经济、更创新的合成路线,促进有机合成和药物发现的发展。
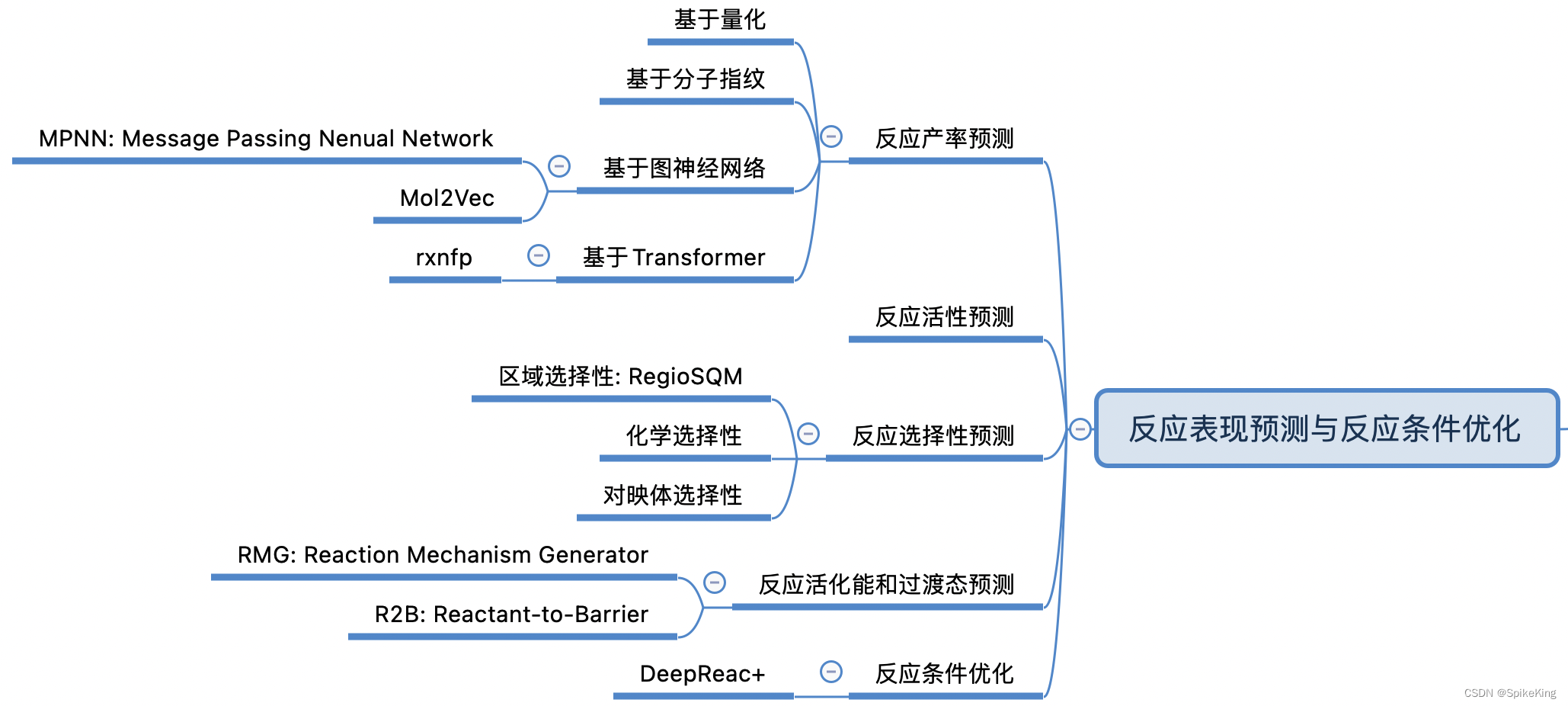
5. 反应表现预测与反应条件优化
反应表现预测与反应条件优化是指利用机器学习等方法,根据给定的反应物和反应类型,预测反应的产率、活性、选择性等性质,以及优化反应的温度、溶剂、催化剂等条件,从而提高反应的效率和经济性。
- 反应产率预测 是指根据反应物的结构和性质,预测反应的产物收率。反应产率预测的方法可以分为基于物理的方法和基于信息的方法。基于物理的方法是利用量子化学计算或分子动力学模拟来模拟反应的过程和机理,从而估计反应的活化能和速率常数,进而计算反应的产率。基于信息的方法是利用机器学习或深度学习的技术,从大量的反应数据中学习反应的隐含规律和特征,从而建立反应物和产率之间的预测模型。
- 反应活性预测 是指根据反应物的结构和性质,预测反应的活化能或速率常数,反映反应的难易程度和速度。反应活性预测的方法可以分为基于物理的方法和基于信息的方法。基于物理的方法是利用量子化学计算或分子动力学模拟来模拟反应的过程和机理,从而计算反应的活化能或速率常数。基于信息的方法是利用机器学习或深度学习的技术,从大量的反应数据中学习反应的隐含规律和特征,从而建立反应物和活性之间的预测模型。⁵
- 反应选择性预测 是指根据反应物的结构和性质,预测反应的立体选择性或化学选择性,反映反应的产物分布和偏好。反应选择性预测的方法可以分为基于物理的方法和基于信息的方法。基于物理的方法是利用量子化学计算或分子动力学模拟来模拟反应的过程和机理,从而分析反应的过渡态和能量差异,进而计算反应的选择性。基于信息的方法是利用机器学习或深度学习的技术,从大量的反应数据中学习反应的隐含规律和特征,从而建立反应物和选择性之间的预测模型。
- 反应活化能和过渡态预测 是指根据反应物的结构和性质,预测反应的活化能和过渡态结构,反映反应的难易程度和机理。反应活化能和过渡态预测的方法可以分为基于物理的方法和基于信息的方法。基于物理的方法是利用量子化学计算或分子动力学模拟来模拟反应的过程和机理,从而计算反应的活化能和过渡态结构。基于信息的方法是利用机器学习或深度学习的技术,从大量的反应数据中学习反应的隐含规律和特征,从而建立反应物和活化能或过渡态之间的预测模型。
- 反应条件优化 是指根据反应物的结构和性质,以及反应的目标,优化反应的温度、溶剂、催化剂、添加剂等条件,从而提高反应的效率和经济性。反应条件优化的方法可以分为基于物理的方法和基于信息的方法。基于物理的方法是利用量子化学计算或分子动力学模拟来模拟反应的过程和机理,从而分析不同条件对反应的影响,进而寻找最佳条件。基于信息的方法是利用机器学习或深度学习的技术,从大量的反应数据中学习反应的隐含规律和特征,从而建立反应物、条件和目标之间的优化模型。




中值滤波)
堆的简单介绍以及实战 heap0)


)






)



