
前情回顾
上次小编为大家讲解了四种以基因组为基础的多组学联合研究方案(基因组与转录组,深入挖掘基因表达信息;基因组联合代谢组与转录组,锁定关键通路;基因组与群体进化,解析物种发展历程;基因组结合GWAS与进化,探索重组遗传效应 ) 并通过八篇高分合作文章作为经典案例进行案例概述。详情可点击→基因组学深入挖掘·研究方案(上篇)进行回看。
相信很多老师根据上期方案概述已经有了适合自己的研究思路或是找到了自己研究中的影子,但是基因组作为一种基础且必要的研究,其身上的挖掘方向远远不止这些~接下来我们将精彩继续,咱们接着谈谈关于基因组学研究的其他几种热点方案~
方案五
基因组与遗传图谱,连锁分析定位性状
技术介绍
遗传图谱是目前最经典的功能基因定位策略,主要基于高通量测序技术开发单核苷酸多态性位点(SNP),并计算多态性标记间的遗传连锁距离,构建高密度遗传图谱,最后结合性状调查对目标区域进行定位。数量性状基因座(quantitative trait locus,QTL)是指基因组中引起数量性状变异的座位。作物的产量、质量、株型、生长发育等大多数重要经济性状及农艺性状通常都为数量性状。利用分子标记,通过连锁分析进行QTL定位,是遗传学中研究数量性状相关功能基因的基本手段和确定分离并分离目标基因的前提。而优质的基因组才能为性状精准定位提供有力支持。
技术路线
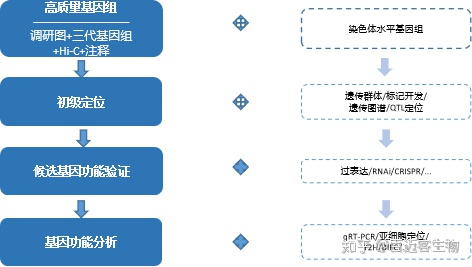
适用范围
双亲杂交得到的性状分离群体
包括暂时性分离群体,如F1、F2、F3、BC等
永久性分离群体,如DH、RIL等
产量、育性、花期、果实性状、抗病虫、抗盐碱、抗寒、品质、株型、性别决定、代谢物等性状定位
高分案例
植物案例一
英文名:The Coix genome provides insights into Panicoideae evolution and papery hull domestication[1]
中文名:薏苡基因组为黍亚科进化及薄皮作物驯化提供新见解
期刊:Molecular Plant(IF:12.084)
合作单位:四川农业大学
主要研究内容:
作者以饲用薏苡 “大黑山”为研究对象,利用PacBio组装了1.62 Gb的薏苡基因组,contig N50达到了2.24 Mb,并通过Hi-C将1.5 Gb的序列定位到10条染色体上。比较基因组分析揭示了薏苡高生物量和生物抗性的可能原因。作者通过对两种不同种壳品种进行转录组差异基因聚类分析,揭示薏苡种壳薄皮形成原因,解析驯化选择机制。最后将两种不同种壳品种杂交产生的F2群体构建遗传图谱,对种壳抗压性、抽穗期、株高和分蘖数等性状QTL定位。后期扩大群体精细定位,进一步将这2个QTLs分别定位到250Kb和140Kb的区间内。该研究有助于推动薏苡关键药用成分的研究和传统中药的现代化应用,为薏苡粮、药、饲专用新品种的高效选育奠定了坚实的基础。
植物案例二
英文名:Population Genomic Analysis and De novo Assembly Reveal the Origin of Weedy Rice as an Evolutionary Game[2]
中文名:群体基因组分析结合从头组装揭示杂草稻作为进化演绎的起源
期刊:Molecular Plant(IF:12.084)
合作单位:沈阳农业大学
主要研究内容:
作者使用WRAH和 Qishanzhan 栽培稻的杂交,获得包含 168 个子代的 RIL 群体,利用 SLAF-seq 技术进行测序,构建高密度遗传图谱。对种子落粒、长芒、高株高、色壳和红果皮5个性状进行定位。并对不同地域品种的333份水稻构建系统发育树,明确WRAH的分群情况,并通过选择性清除分析发现驯化相关基因可能在平行进化中发挥重要作用。基于以上研究作者通过PacBio+Hi-C组装了高质量的亚洲高纬度杂草稻WR04-6基因组,并通过比较基因组分析研究相关基因家族及分化时间。该研究揭示了栽培稻在从野生稻驯化后,近代的遗传改良成为了亚洲高纬度杂草稻与粳型栽培稻遗传趋异的分水岭。
方案六
基因组与表观遗传,解析三维网络调控机制
技术介绍
表观遗传(epigentics) “epi”表示“其上”或“超越”,genetics只设计基因的结构层次,而epigenetics则涉及基因如何发挥其功能以及基因间的互作关系。基因组结合表观调控可更加深入的剖析物种在何时、何地、以何种方式去应用遗传信息的指令。多种表观遗传研究手段间可相互组合,从三维基因、修饰表达、调控元件等多种层面解析不同调控原理。
技术路线
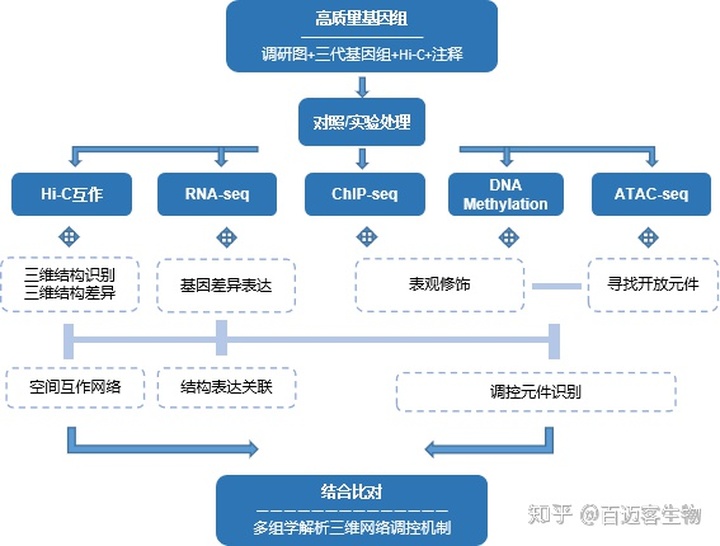
适用范围
不同物种间调控因子差异研究
胁迫等差异环境响应调控机理
发育生长、病理恢复间基因结构/调控机制
不同组织,器官转录活性差异,找到组织特异基因和启动子
非生物逆境,病虫害,营养,激素等处理前后及动物疾病差异
宏观分析细胞在该特定时空下整个基因组的调控网络
高分案例
植物案例
英文名:N6-Methyladenine DNA Methylation in Japonica and Indica Rice Genomes and Its Association with Gene Expression, Plant Development and Stress Responsesence[3] 中文名:粳稻和籼稻基因组N6-甲基腺嘌呤DNA甲基化及其与基因表达、植物生长和应激反应的关系
期刊:Molecular Plant(IF:12.084)
合作单位:中国农业科学院生物技术研究所
主要研究内容:
作者利用PacBio对粳稻日本晴栽培种Japonica Nipponbare (Nip)及籼稻栽培种Indica 93-11进行测序。最终组装Nip基因组大小为380.7 Mb(contig N50=16.97 Mb),93-11基因组大小为396 Mb(contig N50=9.64 Mb)。新组装的Nip和93-11基因组的contigN50分别比先前公布的高2.2倍和460倍。并使用改进的水稻(Nip、93-11)基因组和SMRT测序鉴定了粳稻和籼稻基因组中全基因组的6mA位点的单核苷酸分辨率。并报道了93-11中第一个6mA甲基位点,发现Nip和93-11中6mA位点在着丝粒周围异染色质区域附近富集;6mA与水稻中的活性表达基因有关;Nip、93-11和拟南芥中6mA的分布及其与转录的关系是保守的。6mA与热应激关键基因的表达呈正相关。筛选了与表观遗传学相关的潜在突变体,并发现DDM1对水稻中的6mA修饰是必不可少的。
方案七
种内/种间基因组变异,掌控起源及功能关系
技术介绍
对于大部分动植物,其测序主要基于单一品种作为参考基因组。单一基因组测序通常只能覆盖基因组的80%~90%,且通常只有代表驯化的优良品种的单一基因型能够被准确检测到,因此不同生态型重测序数据一般只有50%~80%能比对到参考基因组上。而通过对两种品系基因组进行denovo(或者与已发表品系进行研究),充分比较其间的基因组变异信息,可以更好的对不同生态型进行表型功能差异分析。结合两种生态型品种杂交子代图谱,将极大的利于后续性状与功能研究。与动物相比植物更具有品种多样性,因此该研究方案目前更多的应用于植物材料中。
而亲缘关系较近的种间材料同样可以分别进行denovo,并通过其间的变异分析掌控物种间的起源进化及功能关系。
技术路线
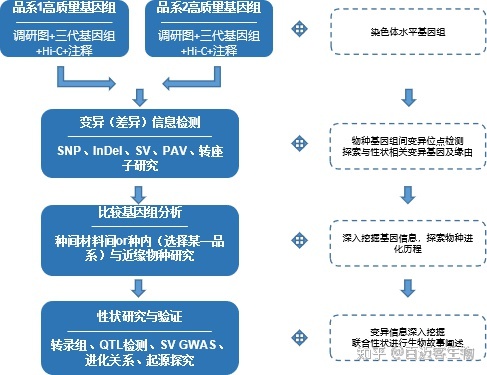
适用范围
品种材料间优秀性状检测与育种
物种间基因组进化关系
大型结构变异与性状分析
高分案例
植物案例一(种间变异)
英文名:Reference genome sequences of two cultivated allotetraploid cottons Gossypium hirsutum and Gossypium barbadense[4] 中文名:异源四倍体陆地棉和海岛棉基因组破译
期刊:Nature Genetics(IF:27.603)
合作单位:华中农业大学
主要研究内容:
作者利用三代测序(PacBio)+光学图谱+Hi-C相结合的方法进行异源四倍体陆地棉和海岛棉基因组组装。组装获得陆地棉Contig L50 = 1.89 Mb,海岛棉Contig L50 = 2.15 Mb,Hi-C染色体挂载效率分别为 98.94%和97.68%。对异源四倍体陆地棉和海岛棉进行全基因组变异分析,包括SNPs和Indels变异分析,染色体结构变异及PAVs分析。将A-亚基因组供体二倍体祖先种亚洲棉G. arboreum与陆地棉和海岛棉进行比较,证明了从二倍体到四倍体多倍化过程中发生了结构变异。通过构建渗入系,将海岛棉的有利染色体片段导入到陆地棉中,并进行QTL定位,在两种代表性种质间探索研究了具有潜在优质纤维质量性状的基因组序列信息,从而帮助棉花改良育种。
植物案例二(种内变异)
英文名:Extensive intraspecific gene order and gene structural variations in upland cotton cultivars[5] 中文名:陆地棉栽培种广泛的基因顺序和基因结构变异
期刊:Nature Communications(IF:12.121)
合作单位:中国农业科学院棉花研究所
主要研究内容:
作者利用三代PacBio测序,对陆地棉(G. hirsutum)遗传研究标准种TM-1和易于转化的生物技术重要品种ZM24 进行测序,组装TM-1基因组2.286 Gb (Contig N50=4.760 Mb), ZM24基因组2.309 Gb(Contig N50=1.976 Mb),利用Hi-C将TM-1组装到染色体水平(挂载率97.4%)。通过二者的基因组与二倍体祖先种之间的比较,发现了大量的遗传变异。TM-1 与 ZM24 比较发现有大量倒位、染色体内易位和染色体间易位,3个最大的结构变异来自于A08 染色体,PAV 基因可能在多倍体的形成过程中已经出现。进一步通过两个栽培种和种质panel的作图群体的单倍型分析显示该区域的重组率受到抑制。本研究为棉花研究领域提供了更多的基因组资源,鉴定的遗传变异,将对未来的棉花育种具有帮助。
方案八
泛基因组,解析物种表型及性状多样性
技术介绍
泛基因组(Pan-genome)即某一物种全部基因的总称,包括核心基因(core gene;由所有个体都存在的基因组成)和非核心基因(dispensable gene;仅在单个个体或部分个体中存在的基因组成)。泛基因组分析有助于理解物种的特征,同时泛基因组图谱提供的基因PAV变异或基因复制等复杂基因组变异,有助于解析作物表型和农艺性状的多样性。选择不同亚种材料进行泛基因组测序,可以研究物种的起源及演化等重要生物学问题;选择野生种和栽培种等不同特性的种质资源进行泛基因组测序,可以发掘重要性状相关的基因资源,为科学育种提供指导;选择不同生态地理类型的种质资源进行泛基因组测序,可以开展物种的适应性进化、外来物种入侵性等热门科学问题。
技术路线
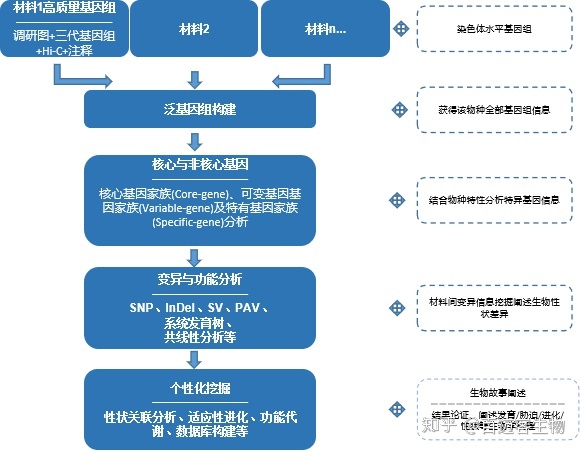
适用范围
多表型作物性状功能研究
功能基因育种基础
多样品种来源传播探索
重要性状基因资源发掘
外来物种入侵考究
高分案例
植物案例一
英文名:The barley pan-genome reveals the hidden legacy of mutation breeding[6] 中文名:大麦泛基因组揭示了突变育种的潜在遗传基础
期刊:Nature(IF:42.778)
研究单位:澳大利亚默多克大学西澳洲大麦联盟
主要研究内容:
作者通过对全球超过2万份大麦种质资源进行遗传多样性分析,最终选出20个能够覆盖大部分大麦遗传多样性的品种(包括地方种、栽培种及野生种)。结合PacBio、Illumina、10x Genomics、Hi-C等技术构建了20个染色体水平基因组,组装基因组大小在3.8Gb-4.5Gb,N50范围为5.0-42.7 Mb。并对不同品种间基因组大片段插入/缺失变异(PAV)、结构变异进行了鉴定。发现大的倒位多态性(>5 Mb)普遍存在,并对经常在优质大麦品种中发现的两个重要的大片段倒位现象进行了分析。该研究首次构建了大麦泛基因组,检测了大量未发现的遗传变异,这些变异将为遗传分析和育种提供有力支撑。
植物案例二
英文名:Multiple wheat genomes reveal global variation in modern breeding[7] 中文名:多个小麦基因组揭示了现代育种中的全面变异
期刊:Nature(IF:42.778)
研究单位:加拿大萨斯喀彻温大学等研究单位
主要研究内容:
作者通过通过NRGene DeNovoMagic3平台结合10X Genomics、Illumina、Hi-C等技术完成了不同地区来源的10个染色体级别和5个scaffold级别的6倍体小麦基因组,组装结果在14.2Gb-14.8Gb。并通过挖掘不同小麦品种间的基因多样性,研究了一些农艺相关性状基因家族。通过基因组比较及ChIP-seq等技术揭示了基因组的转座子TE差异, 外缘染色体片段, 着丝粒倒位以及10个基因组间大的结构变异(SV),这些研究揭示了广泛的结构重排、野生亲缘的引入以及复杂的育种历史导致基因变异,这些育种历史旨在提高小麦对不同环境的适应能力、产量和质量以及抗逆性。基因组间比较研究发现了一个多基因组衍生的富亮氨酸重复结构域的核苷酸结合蛋白库,这种蛋白参与了小麦的抗病性,并且报道了抗虫基因Sm1的基因变异。小麦泛基因组将为发现功能基因和培育下一代现代小麦品种提供理论基础。
动物案例
英文名:Massive gene presence-absence variation shapes an open pan-genome in the Mediterranean mussel[8] 中文名:地中海贻贝的大规模基因存在缺失变异形成了一个开放的泛基因组
期刊: Genome Biology(IF:10.806)
研究单位:意大利里雅斯特大学
主要研究内容:
作者通过Illumina、PacBio等技术构建了1.28G地中海贻贝基因组(Contig N50=71.42 kb)。并对14个地中海贻贝进行高深度重测序,发现其间具有大量的结构变异,大的插入和缺失是地中海贻贝基因组多样性的主要来源。进而将重测序数据与组装基因组比对,并将未比对上的contigs进行组装,构建了1.86Gb的地中海贻贝泛基因组。对泛基因组中的核心与非核心基因组进行分析,富集分析结果表明,非核心基因在胞凋亡、免疫信号传导等与环境适应性相关的通路中显著富集,揭示了地中海贻贝的非核心基因组在增强其环境适应性中的重要作用。
尾声
随着技术的飞速发展,各大杂志编辑们“眼光”越来越高,基因组学研究难以再靠单纯的基因组生信分析独树一帜。如何利用好基因组信息,深入挖掘其中奥义;如何使不同生态型生物学现象得以充分解析;如何使得基因组的数据成为遗传育种、物种保护中的关键信息。种种问题在多组学联合研究的背景下将迎刃而解。当然,小编在这里更多的是列举了目前研究最为热门的几种方案,对于不同物种特性,真正适合的研究方案远不止小编列举的这些,单细胞技术、基因编辑等多种新兴技术正在逐步崛起,后续动植物的研究里也会逐渐细致化,精细化。也期待每一位老师能快速找到适合自己的研究方案,完美的阐述各种生物学故事。
参考文献
[1] Chao G, Yanan W, Aiguo Y et al. The Coix genome provides insights into Panicoideae evolution and papery hull domestication . Mol Plant. 2019.
[2] Sun J, Ma D, Tang L, et al. Population Genomic Analysis and De novo Assembly Reveal the Origin of Weedy Rice as an Evolutionary Game. Mol Plant., 2019.
[3] Zhang Q, Liang Z, Cui X, Ji C et al. N6-MethyladenineDNA Methylation in Japonica and Indica Rice Genomes and Its Associationwith Gene Expression, Plant Development and Stress Responses. Mol Plant., 2018.
[4] M Wang, LTu, D Yuan, et al. Reference genome sequences of two cultivated allotetraploid cottons, Gossypium hirsutum and Gossypium barbadense. Nature Genetics.2019.
[5] Z Yang, X Ge, Z Yang, et al. Extensive intraspecific gene order and gene structural variations in upland cotton cultivars.Nature Communications.2019
[6] Jayakodi M, Padmarasu S,et al. The barley pan-genome reveals the hidden legacy of mutation breeding. Nature.2020
[7] Walkowiak S, Gao L, et al. Multiple wheat genomes reveal global variation in modern breeding.Nature.2020
[8] Gerdol M, Moreira R, et al. Massive gene presence-absence variation shapes an open pan-genome in the Mediterranean musse. Genome Biology.2020


)



)

)










