R包的安装,每次做分析的时候先运行这段代码把R包都安装好了,这段代码不需要任何改动,每次分析直接运行。
options("repos"="https://mirrors.ustc.edu.cn/CRAN/")
if(!require("BiocManager")) install.packages("BiocManager",update = F,ask = F)
options(BioC_mirror="https://mirrors.ustc.edu.cn/bioc/")cran_packages <- c('tidyr','tibble','dplyr','stringr','ggplot2','ggpubr','factoextra','FactoMineR','devtools','cowplot','patchwork','basetheme','paletteer','AnnoProbe','ggthemes','VennDiagram','tinyarray')
Biocductor_packages <- c('GEOquery','hgu133plus2.db','ggnewscale',"limma","impute","GSEABase","GSVA","clusterProfiler","org.Hs.eg.db","preprocessCore","enrichplot")for (pkg in cran_packages){if (! require(pkg,character.only=T,quietly = T) ) {install.packages(pkg,ask = F,update = F)require(pkg,character.only=T) }
}for (pkg in Biocductor_packages){if (! require(pkg,character.only=T,quietly = T) ) {BiocManager::install(pkg,ask = F,update = F)require(pkg,character.only=T) }
}#前面的所有提示和报错都先不要管。主要看这里
for (pkg in c(Biocductor_packages,cran_packages)){require(pkg,character.only=T)
}
#没有任何提示就是成功了,如果有warning xx包不存在,用library检查一下。#library报错,就单独安装。表达矩阵和临床信息的获取、疾病组和健康组的分组工作
rm(list = ls())#首先清空一下环境防止原来数据的影响
#打破下载时间的限制,改前60秒,改后10w秒
options(timeout = 100000)
options(scipen = 20)#不要以科学计数法表示#传统下载方式
library(GEOquery)
eSet = getGEO("GSE57338", destdir = '.', getGPL = F)#这是一个示例数据集,是关于DCM这种病的
#网速太慢,下不下来怎么办
#1.从网页上下载/发链接让别人帮忙下,放在工作目录里
#2.试试geoChina,只能下载2019年前的表达芯片数据
#library(AnnoProbe)
#eSet = geoChina("GSE7305") #选择性代替第8行#前面下载下来的数据保存到了eSet这个变量中
class(eSet)#发现是一个
length(eSet)eSet = eSet[[1]]
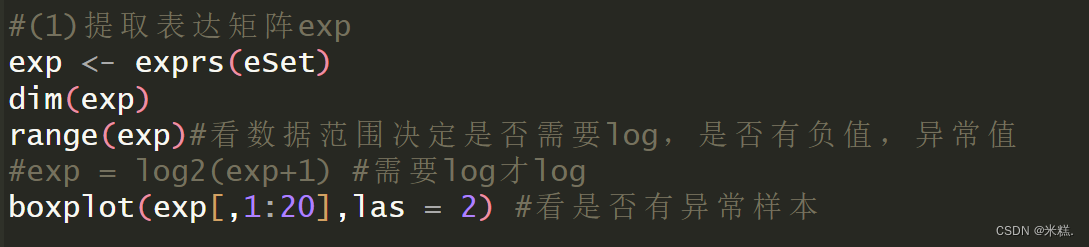
class(eSet)#(1)提取表达矩阵exp
exp <- exprs(eSet)
dim(exp)
range(exp)#看数据范围决定是否需要log,是否有负值,异常值
#exp = log2(exp+1) #需要log才log
boxplot(exp[,1:20],las = 2) #看是否有异常样本#(2)提取临床信息
pd <- pData(eSet)
as.data.frame(pd)
pd$title
k = str_detect(pd$title,"Non|CMP");table(k)
pd<-pd[k,]
#(3)让exp列名与pd的行名顺序完全一致
p = identical(rownames(pd),colnames(exp));p
if(!p) {
#intersect用于找出两个向量间的公共元素,返回值是两个向量中相同的元素
#intersect函数比较的时候是不考虑下标的,只要某个元素在传给该函数的两个向量中同时存在,那这个元素就是函数返回值的某个元素s = intersect(rownames(pd),colnames(exp))exp = exp[,s]#根据名字提取exp中的列pd = pd[s,]#根据名字提取pd的行
}#这样得到的exp列名和pd行名顺序完全一致#(4)提取芯片平台编号,后面要根据它来找探针注释
gpl_number <- eSet@annotation;gpl_number
save(pd,exp,gpl_number,file = "step1output.Rdata")# 原始数据处理的代码,按需学习
# https://mp.weixin.qq.com/s/0g8XkhXM3PndtPd-BUiVgw
eSet = getGEO("GSE57338", destdir = '.', getGPL = F)是我们需要根据情况修改的第一句代码,用哪个数据集就把第一个参数改成哪个数据集。
再来研究一下eSet

getGEO函数下载下来的数据集我们是用eSet这个变量接收的,通过上面一系列代码可以知道这是一个列表,且只有一个元素(如果有两个元素就说明是不同批次的或者同一个数据集是不同公司测的),既然只有一个元素那直接提取出来就是了,提取列表中的元素要用两个中括号,提取出来第一个元素之后覆盖掉eSet中的内容,此时再次查看eSet的类型,发现是一个看不懂的东西,反正也是一个类型,这是一个高级类型,总之就理解成接收了我们下载的GEO数据集的一个对象。这个eSet的内容长这样子

这一大堆乱七八糟的是肯定没法直接扔进后续的函数里面做差异分析的,因此接下来我们要提取表达矩阵,直接运行这段代码就行,这段代码不需要修改
其中把eSet这个对象扔到exprs这个函数里面就能得到对应的表达矩阵了,表达矩阵长这样

其中每一列是一个样本,每一行的行名其实是一个探针,而每一个探针都会对应一个基因,后续我们会根据探针与基因名的对应关系把这些行名改成基因名。现在只需要明确一点就是探针的表达量代表基因的表达量就行。
我们把表达矩阵存到exp里面,dim(exp)用来查看这个矩阵的维度(查看维度要做什么后面再说),range(exp)用来查看这个矩阵中元素的范围,如果数据从个位数到几万都有,那这个矩阵中元素就是没有取过对数的,我们需要手动取一下以2为底的对数,不要问为什么取2的对数不是其他的,这就是一个约定俗成的东西,其中在取对数的这一步我们对exp矩阵的每个元素都做了+1的运算,这个确实会改变基因的表达量,但是我们是做差异分析,基因表达量都+1并不会改变他们之间的差异性,之所以+1是为了防止出现一些负数等等。然后使用基本绘图系统的boxplot函数绘制了一个箱线图,boxplot的第一个参数传了exp这个矩阵的前20列,这是因为我用的这个数据集前面查看维度的时候发现是三万多行,三百多列,直接用exp作为参数绘制箱线图非常的密集而导致难以观察,所以我取了前20行,如果前面dim(exp)的时候发现矩阵的列很少,那就不用取前20列了。我用的这个数据集前20绘制的箱线图长这样

发现数据还是很正常的,没有上下浮动很明显的样本。如果有异常就需要做一些另外的处理比如删除等等,这个以后单独介绍一下,这里就不介绍了。
接下来再提取临床信息
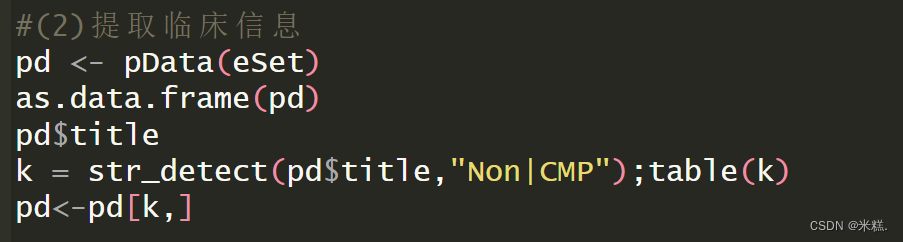
使用pData(eSet)函数就能提取到临床信息了,这个eSet就是我们最开始时用来接收下载的数据的那个对象,函数的返回结果是一个数据框,也就是说临床信息我们使用一个名为pd的数据框保存。pd长这样

可以看到所谓的临床信息包含了title,这一列有特发性扩张型心肌病左心室,缺血性左心室,非衰竭左心室这三种情况,后面还有什么日期等等一系列的临床信息,我想要研究扩张型心肌病和健康人基因表达的差异,那我就根据title这一列中的字符串筛选出这两种情况,也就是把缺血性左心室剔除,我们发现特发性扩张型心肌病左心室这种情况有一个特有的字符串叫做Non,而非衰竭左心室也就是健康样本有一个字符串叫做CMP,运行代码
k = str_detect(pd$title,"Non|CMP");table(k)将会得到一个逻辑向量,如果title这一列(也就是一个向量)的元素中有Non或者CMP,就返回TRUE,否则返回False,再运行代码pd扩张型心肌病和健康样本这两种情况的行了。
接下来要让exp的列名和pd的行名完全一致,至于为什么后面再解释。

identical(rownames(pd),colnames(exp))用于查看pd(只有扩张型心肌病和健康样本这两种情况的数据框)的行名和exp(表达矩阵)的列名 是不是相同,pd的行名和exp的列名都是不同样本名,他们是有可能顺序相同的,如果不同我们执行if语句里面的内容把他们变成顺序相同,intersect函数用于找出两个向量中的公共元素,返回值是一个向量,该向量中的元素均为传给intersect函数的两个参数比如x和y的公共元素,无需考虑下标是不是相同,只要一个元素在x和y两个向量中同时存在,那么这个元素就是intersect返回结果的其中一个元素。我们用s来接收这个intersect函数的结果,这个结果就是一些样本名,然后根据这些样本名提取出exp的列和pd的行并覆盖掉exp和pd,这样就能保证exp的列和pd的行是完全一样的。
预处理还需要把探针转换成对应的基因名,这个代码一般是不需要改动的
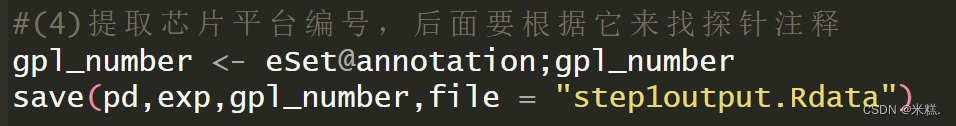
我们看到这里提取eSet中的元素用的是@,这个符号的功能和数据框提取某一列用的那个$是一个道理。一般eSet第一次提取用的是@,后续可能是@也可能是$至于怎么提取可以通过点左上角环境中的这个eSet变量,点开是这样的,根据这里的结构决定是用@还是$提取元素

为了防止代码太长不便于维护,我们把目前所得到的,后续有用的变量存起来
save(pd,exp,gpl_number,file = "step1output.Rdata"),其实目前有用的就是存有临床信息且经过处理之后只剩下扩张型心肌病和健康两种情况的那个数据框pd,以及表达矩阵exp,还有刚才提取到的芯片平台编号,这个编号用于后续把探针转换成基因名。
# Group(实验分组)和ids(探针注释)
rm(list = ls())
load(file = "step1output.Rdata")
# 1.Group----
library(stringr)
# 标准流程代码是二分组,多分组数据的分析后面另讲# 使用字符串处理的函数获取分组k = str_detect(pd$title,"Non");table(k)
# 需要把Group转换成因子,并设置参考水平,指定levels,对照组在前,处理组在后
Group = ifelse(k,"healthy","DCM")
table(Group)
#这个数据框是为了检查一下分组是不是成功
check_df<-data.frame(pd$title,Group)
#把Group这个向量转换成因子类型,前面是对照组
Group<-factor(Group,levels = c("healthy","DCM"))
#2.探针注释的获取-----------------
#捷径
library(tinyarray)
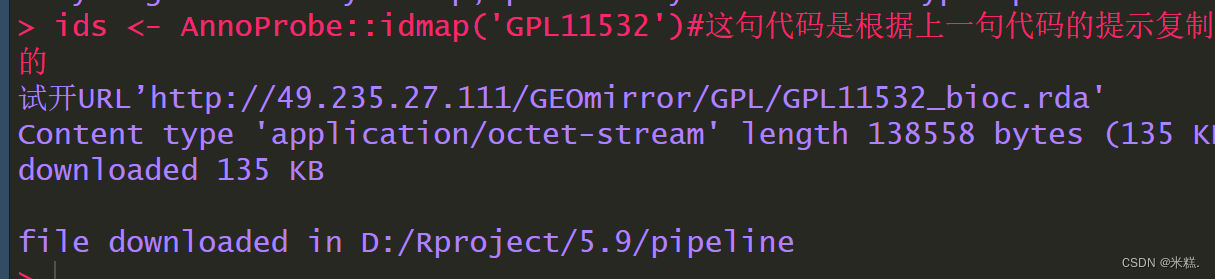
find_anno(gpl_number) #辅助写出找注释的代码
ids <- AnnoProbe::idmap('GPL11532')#这句代码是根据上一句代码的提示复制来的
#如果能打出代码就不需要再管其他方法。
#如果使用复制下来的AnnoProbe::idmap('xxx')代码发现报错了,请注意尝试不同的type参数
#如果不知道type能取什么,可以使用?idmap来查看
#如果显示no annotation avliable in Bioconductor and AnnoProbe则要去GEO网页上看GPL表格里找啦。#捷径里面包含了全部的R包、一部分表格、一部分自主注释
#方法1 BioconductorR包(最常用,已全部收入find_anno里面,不用看啦)
if(F){gpl_number #看看编号是多少#http://www.bio-info-trainee.com/1399.html #在这里搜索,找到对应的R包library(hgu133plus2.db)ls("package:hgu133plus2.db") #列出R包里都有啥ids <- toTable(hgu133plus2SYMBOL) #把R包里的注释表格变成数据框
}
# 方法2 读取GPL网页的表格文件,按列取子集
##https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GPL570
# 方法3 官网下载注释文件并读取
# 方法4 自主注释,了解一下
#https://mp.weixin.qq.com/s/mrtjpN8yDKUdCSvSUuUwcA
save(exp,Group,ids,file = "step2output.Rdata")
重新开一个R脚本清空原来环境中的变量以防止以前运行的内容对本次要运行的代码造成影响
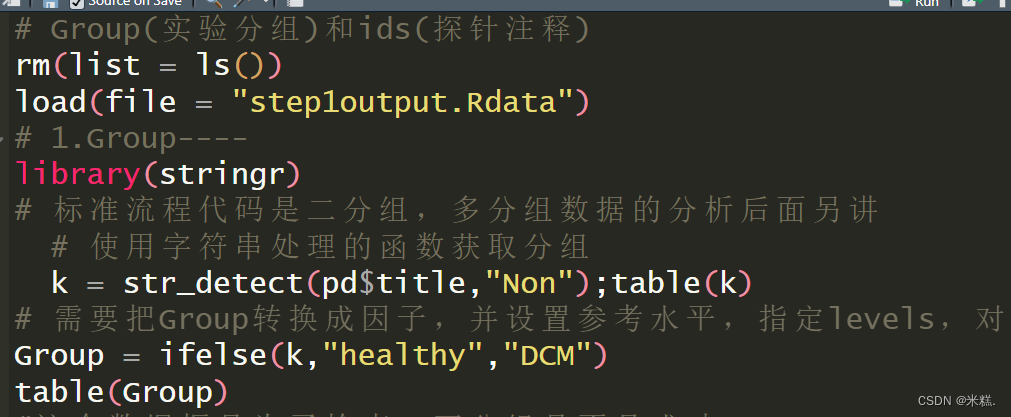
使用r包stringr中的函数str_detect根据title这一列中是否含有Non这个字符串进行筛选,得到一个逻辑向量k,结果如图

这个结果显示有136个健康样本,82个扩张型心肌病样本。
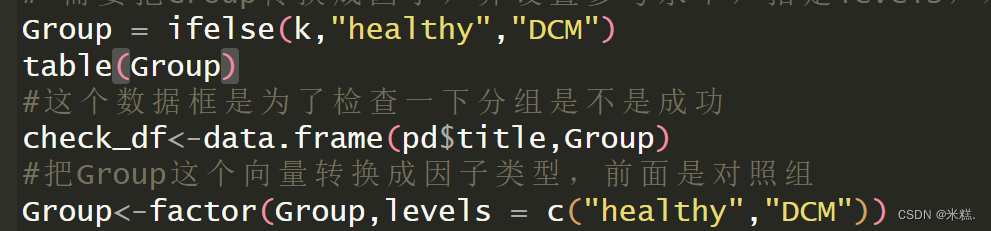
创建一个名为Group的向量,并根据前面的把这个向量的内容改成healthy和DCM(DCM是扩张型心肌病的简称),结果应该是有原本136个TRUE的位置元素被改成了healthy,82个FALSE对应位置的元素被改成DCM。
然后紧接着创建了一个名为check_df的数据框把title这一列和我们刚才创建的Group这个向量放在了一起,顾名思义这个数据框就是为了让我们直观地检查一下是不是正确的分组了。我在这里展示部分check_df的结果

从结果来看我们确实正确的分组了。知道Group这个变量是对的之后,我们就把他转换成因子类型,这是因为后续分析所用到的函数的参数要求。因子这种类型特别适用于处理一些有很多重复元素的向量。

上面这三句代码是在进行探针转换为基因名的工作,find_anno是R包tinyarray中的一个函数,他需要的参数是我们前面得到的编号,扔进去之后会得到一个提示性的结果,我这里是这样

根据这个代码运行结果的提示写了第三句代码






Tkinter笔记(三):控件的定位(1))
、322. 零钱兑换、279.完全平方数)
销售数据分析)






)



