编辑 | 白菜叶
机器学习技术已广泛应用于化学、物理、生物学和材料科学的许多领域。最富有成果的应用之一是从离散量子化学数据中学习势能或相关电子特性的复杂多维函数。特别是,大量努力致力于开发各种原子神经网络 (AtNN) 表示,这些表示是指将目标物理量表示为由原子 NN 表示的原子分量之和的一系列方法。这类方法不仅完全保留了系统的物理对称性,而且相对于系统的大小呈线性缩放,从而能够在复杂系统甚至跨相的多个可变大小系统中进行准确有效的化学动力学和光谱模拟。
在这里,中国科学技术大学蒋彬教授团队讨论了开发高效和可表示的 AtNN 势的不同策略,以及推广这些标量 AtNN 模型以学习具有正确旋转等方差的矢量和张量。研究人员还回顾了生成实用 AtNN 模型的主动学习算法,并展示了 AtNN 在气体表面系统中应用的精选示例,以证明它们准确表示分子系统和凝聚相系统的能力。研究人员指出了在更多应用场景中进一步开发更可靠、可转移和可扩展的 AtNN 表示的众多挑战。
该研究以「Atomistic neural network representations for chemical dynamics simulations of molecular, condensed phase, and interfacial systems: Efficiency, representability, and generalization」为题,于 2022 年 11 月 16 日发布在《WIREs Computational Molecular Science》。

Born-Oppenheimer 近似可以解决静止原子核的电子薛定谔方程,这构成了量子化学的基础,自然产生了(绝热)势能面 (PES) 的概念。一旦 PES 已知,方程的核运动就可以根据系统的大小在化学动力学模拟中以经典或量子力学的方式求解。解决电子结构问题同样可以得出系统的其他电子特性,例如电荷密度、偶极矩、极化率以及不同电子态之间的跃迁和耦合量。虽然势能(和相关特性)可以直接连同分子动力学 (MD) 经典轨迹一起实时计算,但这种从头算分子动力学 (AIMD) 模拟只能使用廉价的电子结构方法,例如密度泛函理论 (DFT),难以研究稀有事件和长时间事件。此外,这种即时方法通常不适用于量子动力学,因为波包是非局部的。因此,更希望使用足够准确和有效的表示来替换动力学模拟中任意核位置的从头算计算,即使对表示参数化的初始投资可能很大。多年来,开发此类表示一直是原子级化学动力学和光谱模拟的关键任务。
获取 PES 的传统方法在很大程度上取决于系统大小及其使用目的。对于具有大量自由度 (DOF) 的复杂系统,例如,凝聚相材料和生物分子,PES 需要具有良好的可转移性和高效率,因此最常使用编码一些基本量子力学概念的(反应)经验力场(EFF)。相比之下,对于气相或气体表面界面的小型系统,各种线性或非线性拟合/插值方法在尽可能高的水平上忠实地表示从头算能量方面取得了巨大成功。这些 PES 可用于描述给定系统的特定过程,但通常不能转移和扩展到其他系统。
在过去的一两年中,现代机器学习 (ML) 技术彻底改变了在广泛的系统中开发势能和其他属性表示的方式。这种基于神经网络和基于内核的回归等方法与那些不依赖物理近似的传统数学表达式有一些相似之处。但是,前者在高维问题上通常具有更高的灵活性和可扩展性。各种强大的基于 ML 的方法已广泛应用于小分子和反应、反应散射、激发态、光谱学和扩展系统。一个特别成功的方法家族基于由 Behler 和 Parrinello 开创的原子神经网络 (AtNN) 框架,它可以跨阶段对小型和大型系统进行统一表示,并在表示分子特性方面取得重大进展。
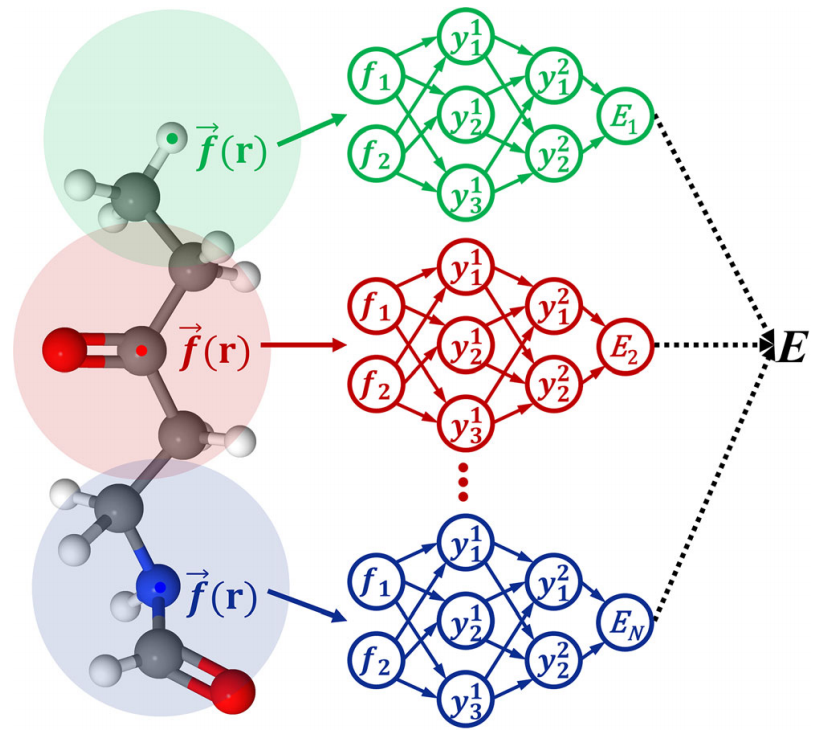
图示:标量(即势能)的一般 AtNN 架构示意图。(来源:论文)
在这里,中科大的研究团队将回顾了这些 AtNN 模型近年来在物理概念和实际实施中的演变情况。他们论文里重点关注最近在提高 AtNN 表示的效率、可表示性和泛化方面所做的研究,以及为特定系统构建可用模型的数据采样方案。讨论了在气体表面系统中选定的 AtNN 应用,展示了它同样能够很好地描述分子中的定向相互作用以及凝聚相的更各向同性和周期性相互作用的能力。
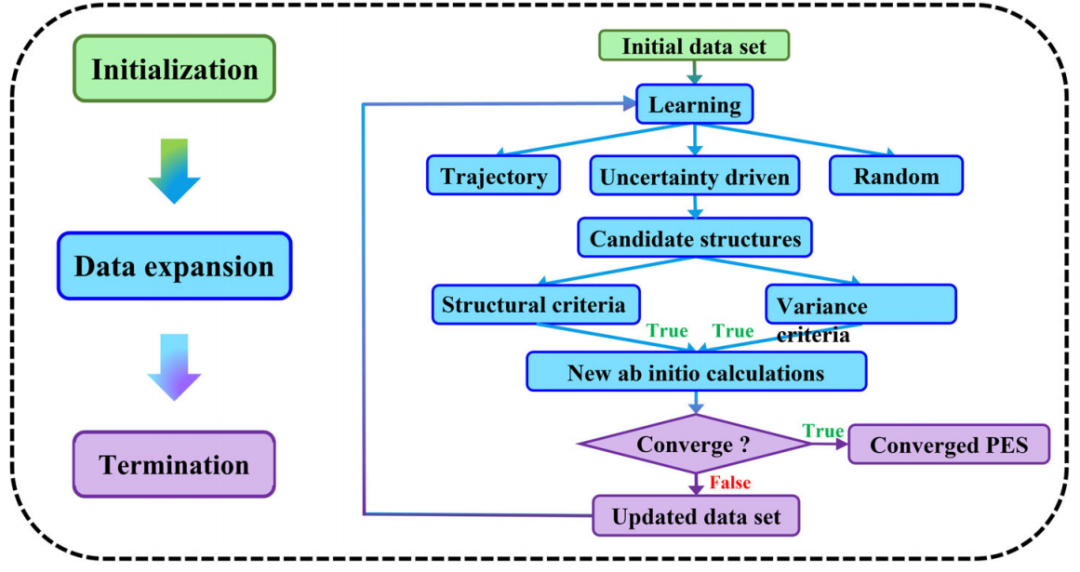
图示:主动学习算法的通用协议。(来源:论文)
尽管取得了众多成功,但 AtNN 表示的未来发展仍然面临一些挑战。首先,很难兼顾 AtNN 表示的效率和可表示性。虽然基于角度特征的 MPNN 模型,例如 REANN,可以通过增加消息传递的数量来包含具有线性缩放的高阶相关性,但仍有提高其效率的空间。特别是,它的并行化不如传统的 AtNN 模型有效,因为现在不同的原子特征是相关的。还提出了更有效的算法来加速高阶交互的直接计算,例如,在 NICE 模型中。更多的数值测试将非常有助于确定实际应用中效率和可表示性之间的最佳平衡。
其次,原子框架本质上依赖于局部近似,通过这种近似,每个原子组件只考虑局部相互作用。由于结合了原子环境内外原子之间的相关性,已经发现 MPNN 模型可以描述一些非局部相互作用。然而,有人认为 MPNN 模型不能像直接增加截止距离那样有效地描述长程相互作用。此外,在一些特殊的分子中,例如CnH4,长程相互作用衰减非常缓慢,并且任何已发布的 AtNN 模型似乎都无法捕捉到端氢旋转时势能的变化。最近的一项工作表明,可以通过以自我注意的方式传递球谐函数来合并非局部几何校正来克服这个问题。为了改进 AtNN 模型的远程行为,库仑相互作用已添加到基于 AtNN 学习的原子电荷的势能中。此外,还引入了电荷平衡方案来描述远程电荷转移。预计将沿着这个方向开展更多工作。
第三,虽然为给定系统建立 AtNN 模型的主动学习协议已经建立,但它们远不是复杂系统的黑盒工具。在大型系统中,需要更明智的方法来衡量预测方差和整个配置之间的相似性,以实现更好的数据效率。另一方面,添加新点的迭代过程需要重复训练 AtNN 模型,这成为主动学习算法的计算瓶颈,需要最小化。此外,尽管一些努力很有希望,但在大型化学空间中从头收集参考数据以生成一系列不同大小和组成的系统的 AtNN 模型仍然非常具有挑战性。
最后,将 AtNN 表示进一步推广到更复杂的场景将很有趣,例如,在存在外部场甚至分子尺度的受限场的情况下。在这种情况下,势能和相关电子特性将受到额外的对称性约束。另一方面,通过求解相应的薛定谔方程直接构建电子和核波函数的 AtNN 表示也很有前途。总而言之,有理由期待 AtNN 表示在未来几十年的各个方面会有更爆炸性的发展。
论文链接:https://wires.onlinelibrary.wiley.com/doi/10.1002/wcms.1645
相关报道:https://phys.org/news/2022-12-breakthroughs-atomistic-neural-network-representations.html
人工智能 × [ 生物 神经科学 数学 物理 材料 ]
「ScienceAI」关注人工智能与其他前沿技术及基础科学的交叉研究与融合发展。
欢迎关注标星,并点击右下角点赞和在看。
点击阅读原文,加入专业从业者社区,以获得更多交流合作机会及服务。





)








)

学习总结)


