本有解决标题中的两个问题
1.答案是修改不了,不如重新制作一个seurat对象。
试图使用rownames(obj)=featurenames是不成功的
记录 客户需求遇到一个问题:作者提供的rds文件行名为ensemble id,如何改成gene symbol。
作者提供的rds文件如下所示
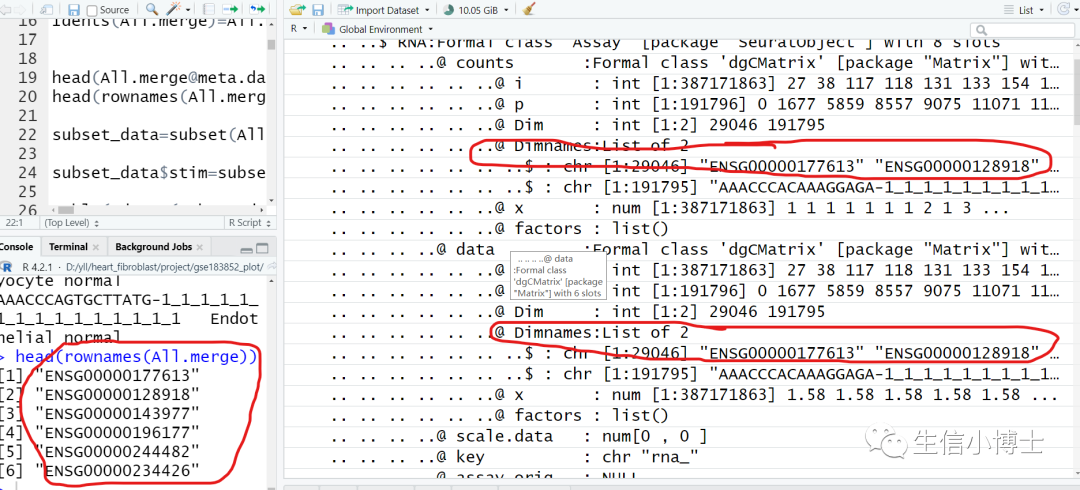
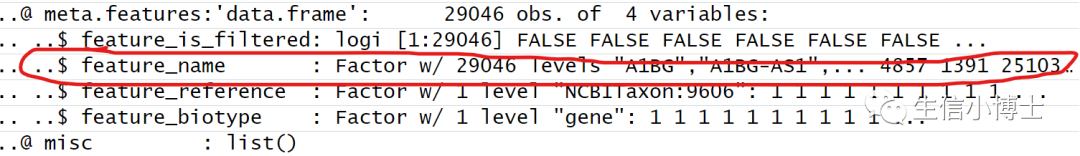
同时发现,作者也提供了相对应的gene symbol
如果直接使用作者提供的rds文件画图的话
FeaturePlot(All.merge,features = "ENSG00000177613")
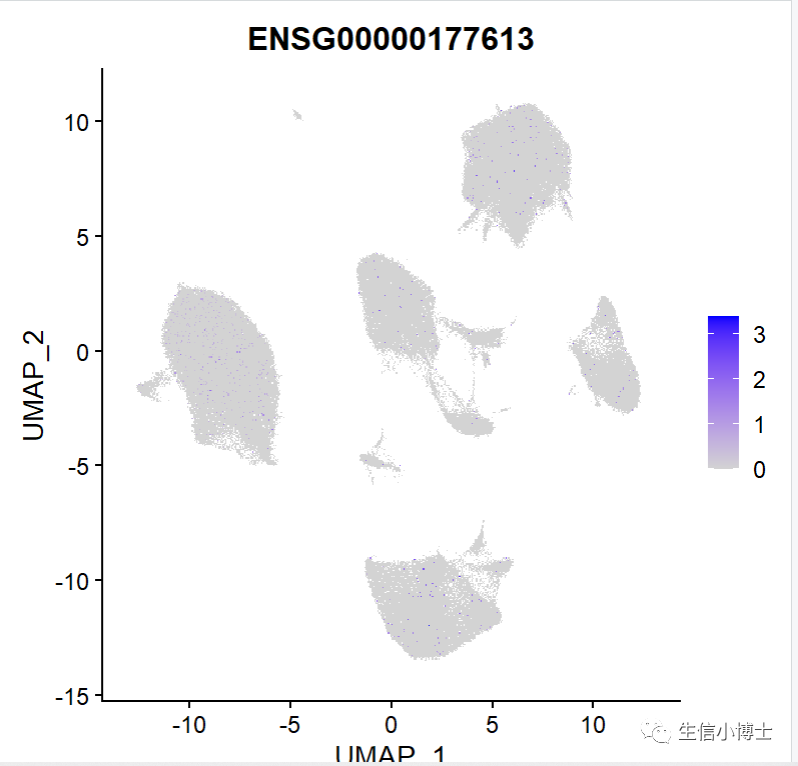
如何把行名改为gene symbol呢?
去github发现,有人提供了一个RenameGenesSeurat函数
#https://github.com/satijalab/seurat/issues/757
#https://github.com/satijalab/seurat/issues/235
仔细阅读该函数,发现本质就是把counts@Dimnames[[1]] 和data@Dimnames[[1]] 还应该有scale.data@Dimnames[[1]] 的名字改为gennesymbol
这里我把scale.data@Dimnames[[1]]注释掉,是因为我这个rds文件中的scale.data为空值,否则会报错的
RenameGenesSeurat <- function(obj = ls.Seurat[[i]], newnames = HGNC.updated[[i]]$Suggested.Symbol) { # Replace gene names in different slots of a Seurat object. Run this before integration. Run this before integration. It only changes obj@assays$RNA@counts, @data and @scale.data.print("Run this before integration. It only changes obj@assays$RNA@counts, @data and @scale.data.")RNA <- obj@assays$RNAif (nrow(RNA) == length(newnames)) {print("ensemble_id length=genesybol length")if (length(RNA@counts)) RNA@counts@Dimnames[[1]] <- newnamesif (length(RNA@data)) RNA@data@Dimnames[[1]] <- newnames# if (length(RNA@scale.data)) RNA@scale.data@Dimnames[[1]] <- newnames} else {"Unequal gene sets: nrow(RNA) != nrow(newnames)"}obj@assays$RNA <- RNAreturn(obj)}subset_data=RenameGenesSeurat(obj = subset_data,newnames = subset_data@assays$RNA@meta.features$feature_name)
这样的话就把gene symbol改成功了
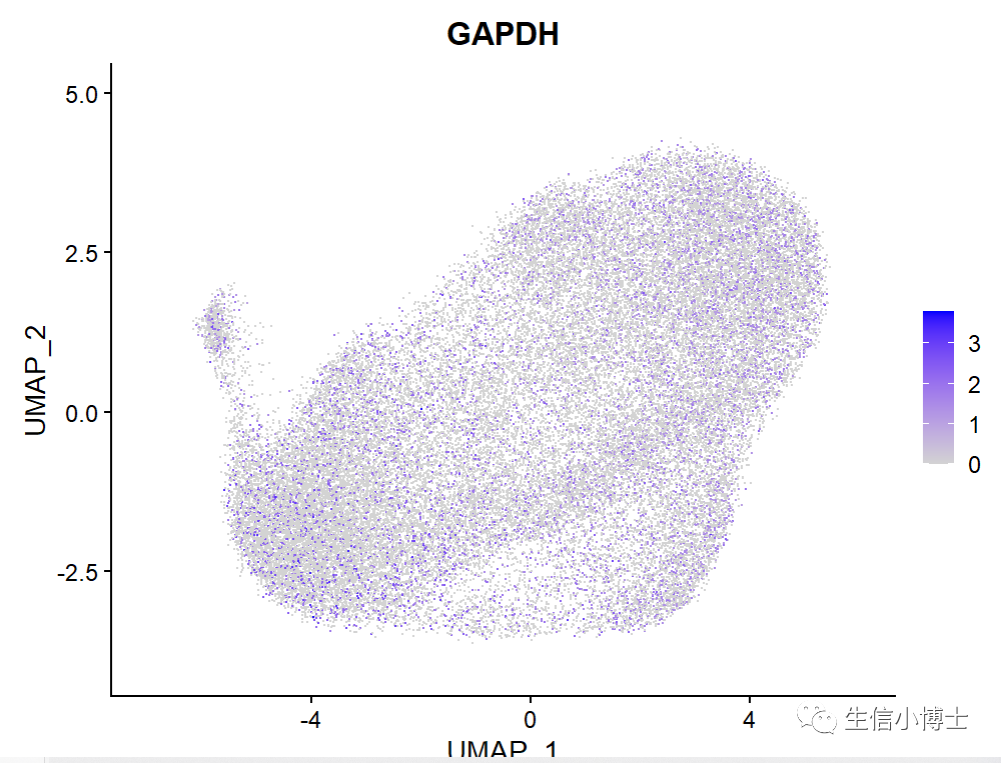
但是
如果你运行FindMarkers或者findallmakers函数的话,还是会报错的。应该是因为FindMarkers运行时候,需要使用rownames,而我们的rownames此时在不同的slot中可能是不一样的。
所以呀
对于这种rds文件,开始先别改行名,全部用自带的ensemble id。可视化的时候再使用RenameGenesSeurat函数修改基因。
否则,会一直报错到怀疑人生。
其实,最好的办法,可能是把count提取出来,自己制作rds文件,这样就不会有各种报错啦。
在最开始的时候,就把可能报错的小火苗掐灭。

==================
FeaturePlot个性化需求,如何把所有阳性表达的spot放到图的前面
常规画图的话Seurat::FeaturePlot(object=All.merge, features="GAPDH",label = T,raster=FALSE,repel = T,split.by = "group" )
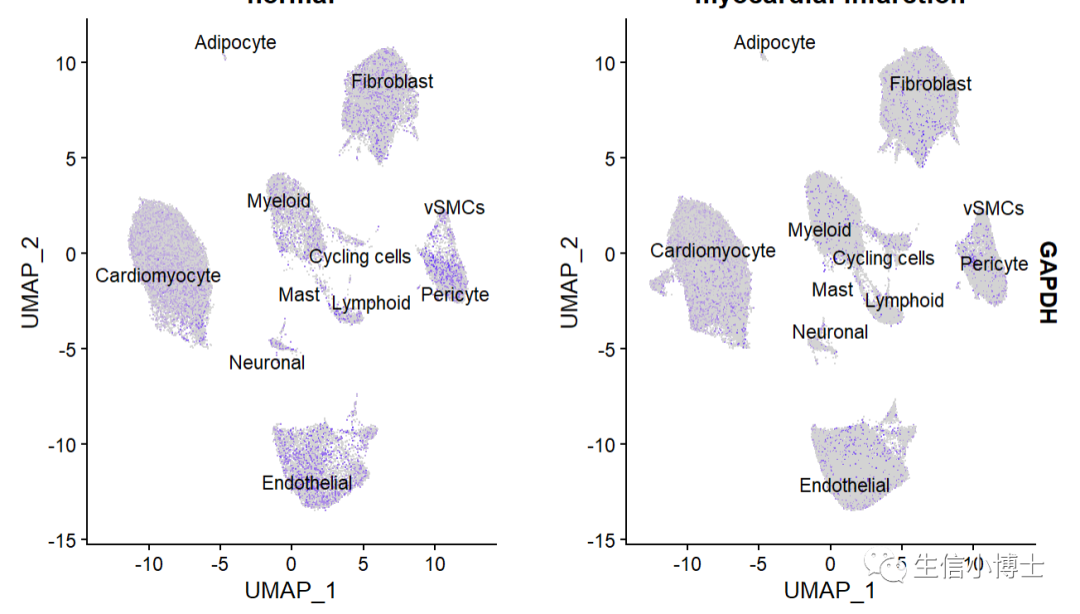
ACTB好像齐一点???

所有阳性表达的spot放到图的前面,结果和方法如下,其实就是对细胞进行重新排个序
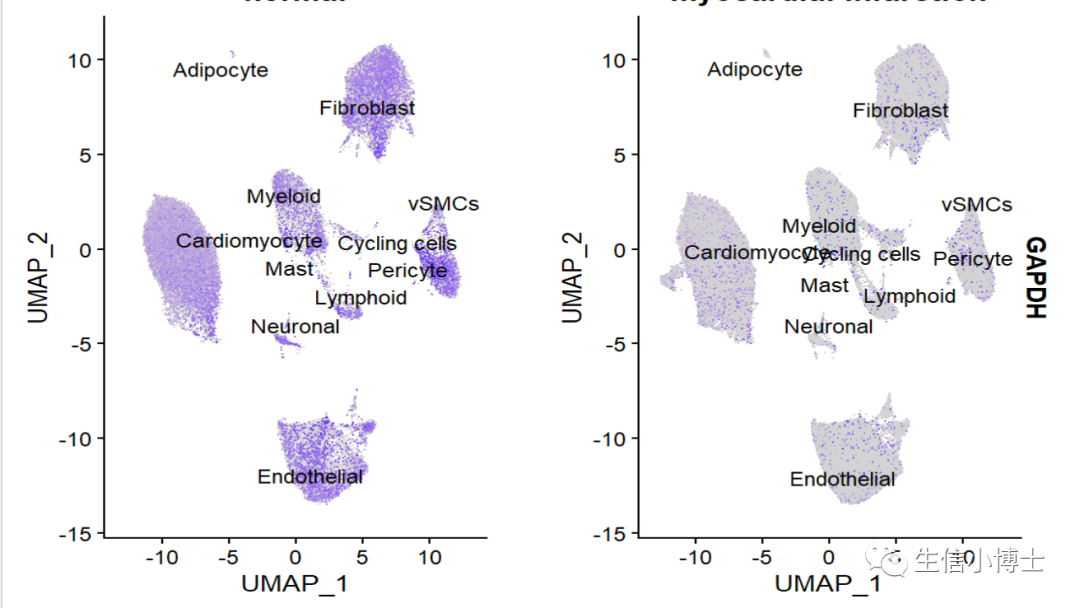
#https://github.com/satijalab/seurat/issues/757#https://github.com/satijalab/seurat/issues/235##################gene plot_bring the postitive dots to the fronts# First, the usual command to make the feature plots and return the ggplot2 objectsfp <- Seurat::FeaturePlot(object=All.merge, features="GAPDH",label = T,raster=FALSE,repel = T,split.by = "group" )fp[[1]]$data$GAPDH# Here, fp is a list of objects, one per feature plot# Then, re-order the "data" dataframe in increasing order of gene expressionsfor (i in 1){fp[[i]]$data <- fp[[i]]$data[order(fp[[i]]$data$GAPDH),]}fp可以看到两个方法 视觉差异还是挺大的






 算法(附完整实现代码))




)







初识对象——C++中的string)