BAM文件时存储比对数据的常用格式,可用于短reads和长reads数据。BAM是二进制压缩格式,SAM文件为其纯文本格式,CRAM为BAM的高压缩格式,IO效率相比于BAM略差,但是占用存储空间更小。
1. BAM文件的比对信息
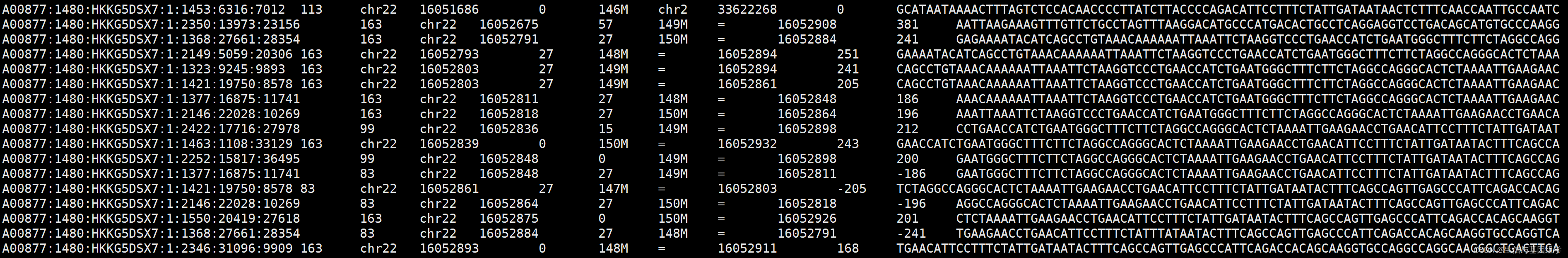
BAM的核心信息-record(也叫alignment section,比对信息), 记录序列比对内容,每一行都是一条read比对信息,各部分内容如下图所示:
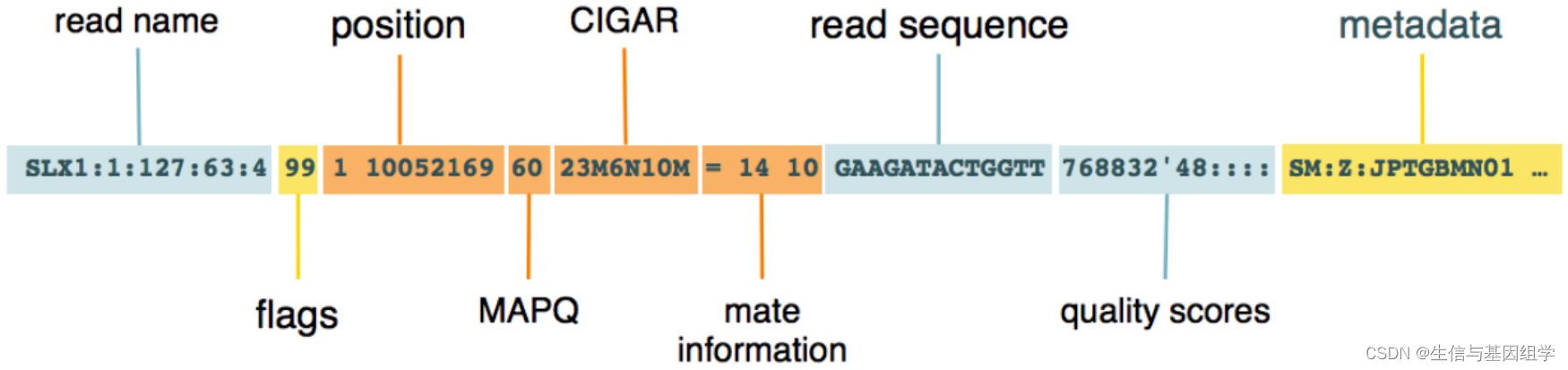 比对信息各部分内容含义:
比对信息各部分内容含义:
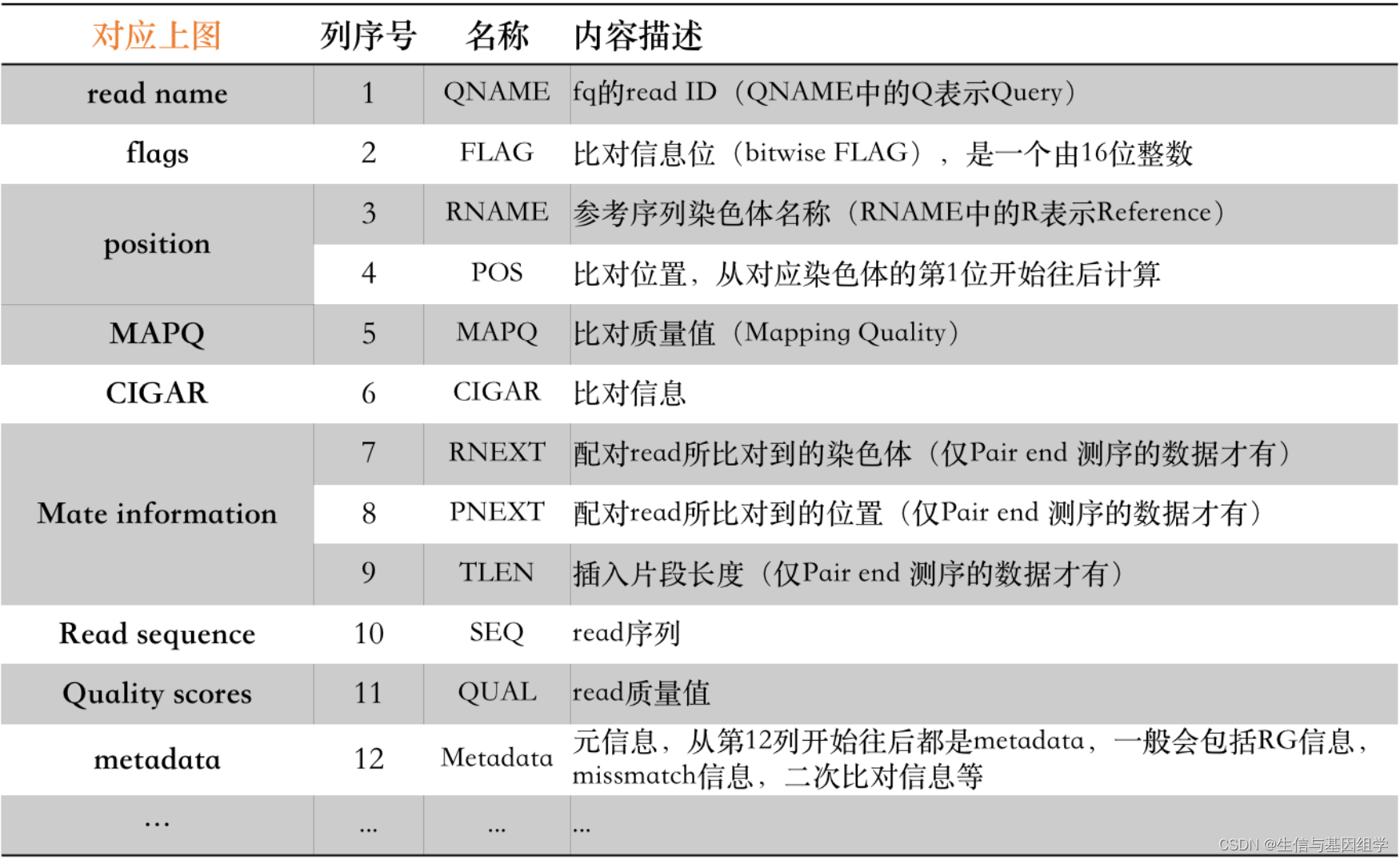
2. 比对信息关键内容 FLAG和CIGAR
2.1 FLAG
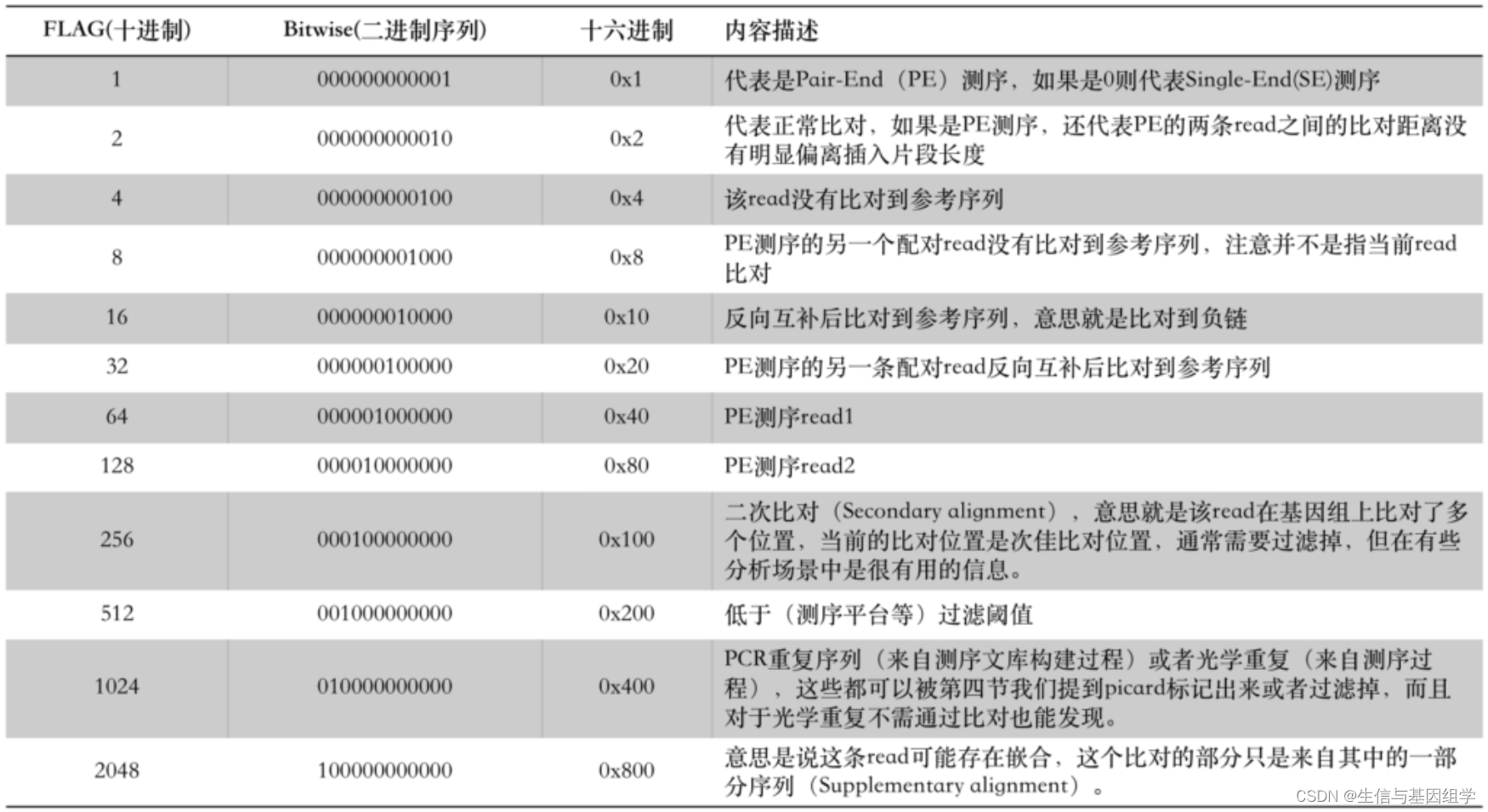
FLAG记录了read的比对情况,FLAG信息使用数字来表示比对结果,比如77,163等,其中77 = 1 + 4 + 8 + 64, 163 = 1 + 2 + 32 + 128, 其中相加使用的数据含义如下表所示:
比如163数据可以理解为:
PE测序reads + 正常比对上参考序列 + 另一条配对reads反向互补后比对到参考序列 + 该序列是read2
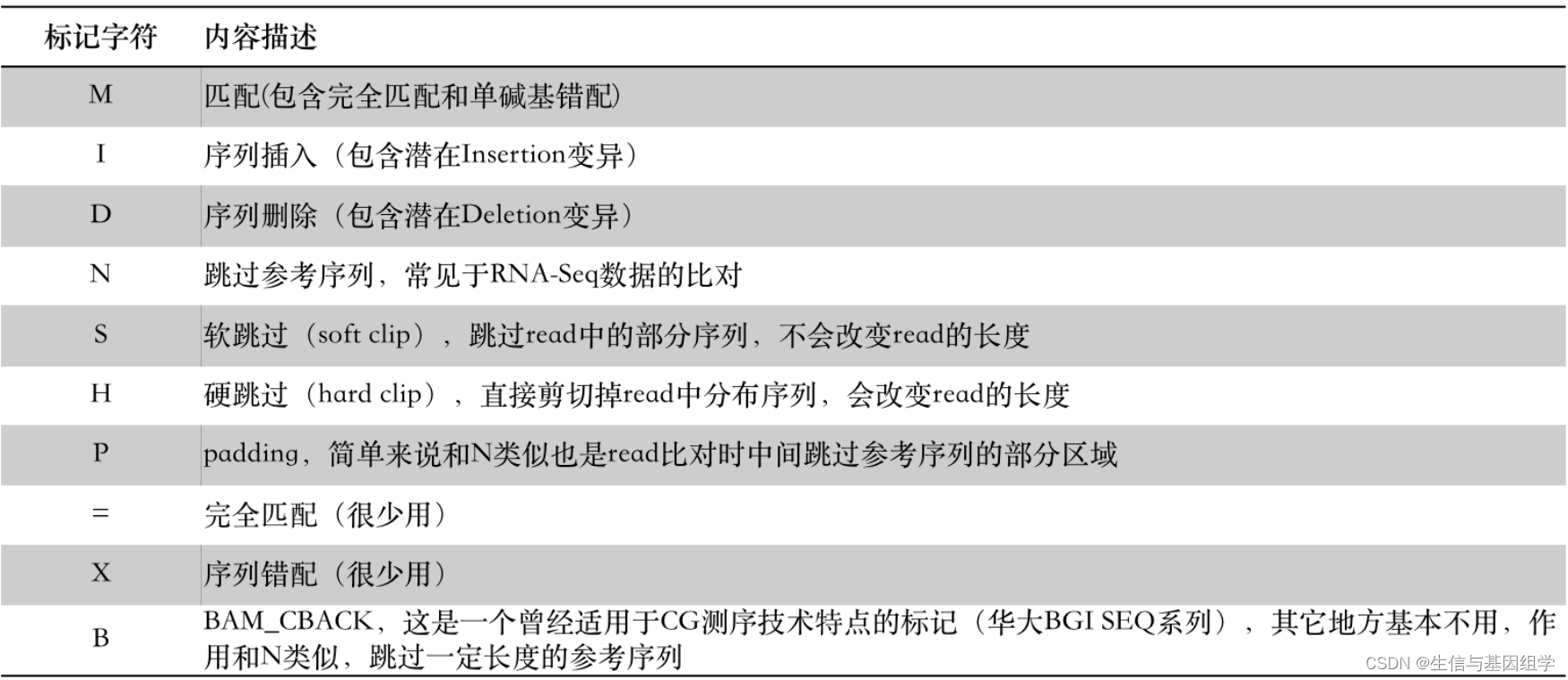
2.2 CIGAR
CIGAR全称Compact Idiosyncratic Gapped Alignment Report, 称为“雪茄”字符串。CIAGR相比对FLAG更容易理解,使用数字+字母组合表示reads的比对情况,比如,一条150bp长的read比对到基因组之后,假如CIGAR字符串内容为:33S117M,其意思是说在比对的时候这条read开头的33bp在被跳过了(S),紧接其后的117bp则比对上了参考序列(M), 其中S代表软跳过(Soft clip),M代表匹配(Match)。
CIGAR标记字符共有10个: “MIDNSHP=XB”,具体分别代表read比对时的情况如下图所示:
特别注意M标记字符,其并非100%全部匹配,单碱基的错配也会标记为M。
3. samtools view查看指定BAM文件指定区域
对于BAM文件的查看可使用samtools。
# 生成索引文件
samtools index sample.sorted.bam# 跳转到chr22染色体
samtools view sample.sorted.bam chr22 # 跳转到chr22:16050000位置
samtools view sample.sorted.bam chr22:16050000# 跳转到chr22:16050000-16060000位置
samtools view sample.sorted.bam chr22:16050000-16060000
4. samtools view提取BAM文件指定染色体区域
对于bam文件过大,可以提取指定区域并生成小的bam文件,导出后再IGV进行查看。
# 提起chr22区域,并输出为bam格式
# —h 表示包含header信息
samtools view -h sample.sorted.bam chr22:16050000-16060000|\
samtools view -Sb - > chr22.region.bam# 查看bam
samtools view chr22.region.bam|less -S
生信分析进阶文章推荐
生信分析进阶1 - HLA分析的HLA区域reads提取及bam转换fastq
生信分析进阶2 - 利用GC含量的Loess回归矫正reads数量
生信分析进阶3 - pysam操作bam文件统计unique reads和mapped reads高级技巧合辑






函数(将多个可迭代对象的元素配对,创建一个元组的迭代器)zip_longest())
 GPT模型详解)
![[数据集][目标检测][数据集][目标检测]智能手机检测数据集VOC格式5447张](http://pic.xiahunao.cn/[数据集][目标检测][数据集][目标检测]智能手机检测数据集VOC格式5447张)
)



)

)



