本系列持续更新Seurat单细胞分析教程,欢迎关注!
维度确定
为了克服 scRNA-seq 数据的任何单个特征中广泛的技术噪音,Seurat 根据 PCA 分数对细胞进行聚类,每个 PC 本质上代表一个“元特征”,它结合了相关特征集的信息。因此,顶部主成分代表了数据集的稳健压缩。但是,我们应该选择包含多少个成分? 10? 20? 100?
在 Macosko 等人中,我们实施了受 JackStraw 程序启发的重采样测试。虽然 Seurat 中仍然可用,但这是一个缓慢且计算成本高昂的过程,并且我们不再用于单细胞分析。
另一种启发式方法生成“Elbow plot”:根据每个主成分解释的方差百分比对主成分进行排名(ElbowPlot() 函数)。在此示例中,我们可以观察到 PC9-10 周围有一个“Elbow”,这表明大部分真实信号是在前 10 个 PC 中捕获的。
ElbowPlot(pbmc)
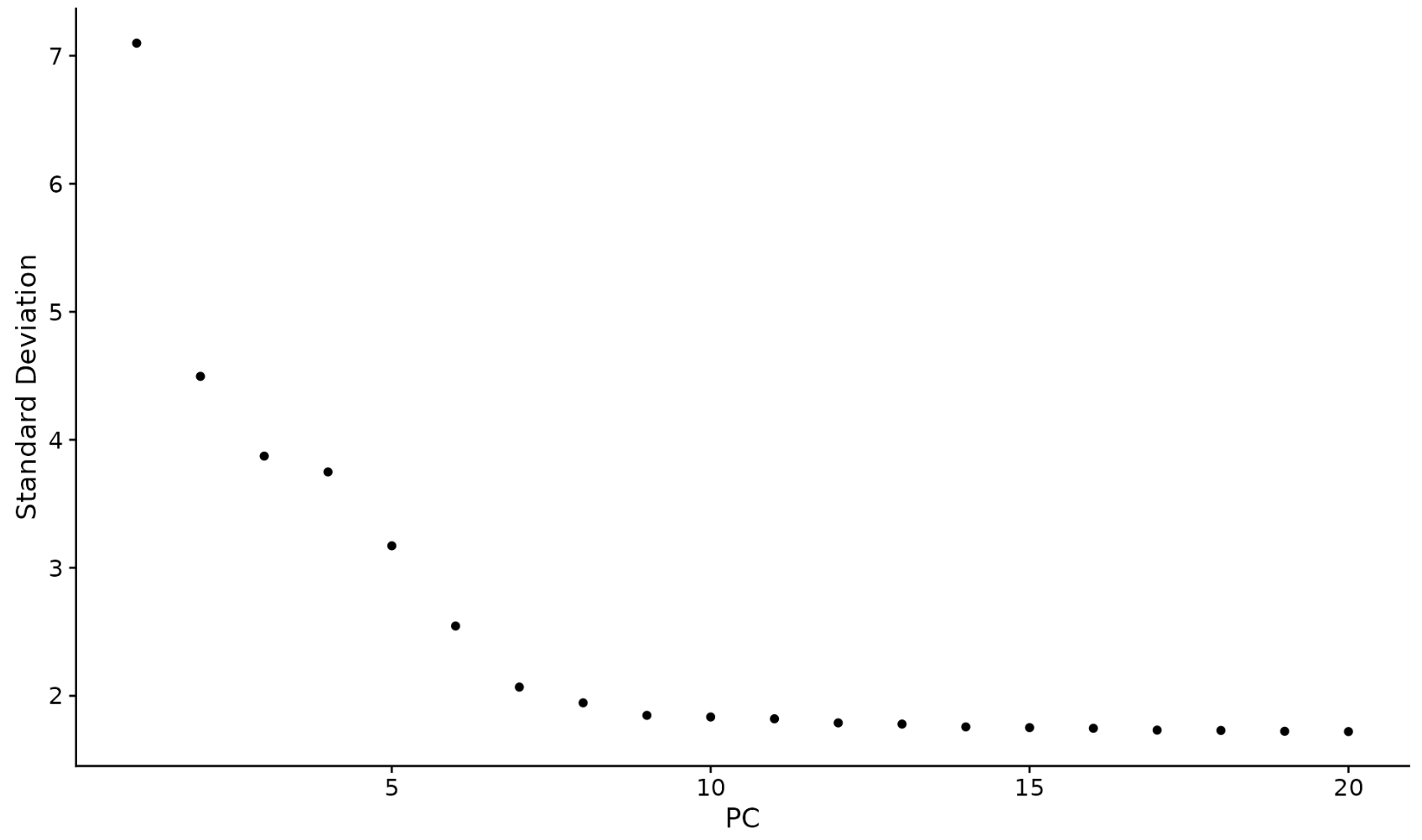
识别数据集的真实维度——对于用户来说可能具有挑战性/不确定性。因此,我们向用户建议采用多种方法。第一个是更多的监督,探索 PC 以确定异质性的相关来源,并且可以与 GSEA 等结合使用。第二个(ElbowPlot) 第三个是常用的启发式,可以立即计算。
在此示例中,我们可能选择 PC 7-12 之间的任何值作为截止值。
我们在这里选择了 10 个,但鼓励用户考虑以下事项:
-
树突状细胞和 NK 与 PC 12 和 13 密切相关的基因定义了罕见的免疫子集(即 MZB1 是浆细胞样 DC 的标记)。然而,这些组非常罕见,在没有先验知识的情况下,很难将它们与这种大小的数据集的背景噪声区分开来。 -
鼓励用户使用不同数量的 PC(10、15,甚至 50!)重复下游分析。 -
用户在选择该参数时偏高。例如,使用 5 PCs 执行下游分析会对结果产生重大不利影响,这将提醒用户重新分析与思考。
细胞聚类
Seurat 应用基于图的聚类方法,以(Macosko 等人)中的初始策略为基础。重要的是,驱动聚类分析(基于先前识别的 PC)的距离度量保持不变。然而,我们将细胞距离矩阵划分为簇的方法已得到显着改进。
Seurat 的方法深受最近手稿的启发,该手稿将基于图的聚类方法应用于 scRNA-seq 数据和 CyTOF 数据 。简而言之,这些方法将cell嵌入到图结构中 - 例如 K 最近邻 (KNN) 图,在具有相似特征表达模式的cell之间绘制边缘,然后尝试将该图划分为高度互连的quasi-cliques’ 或 ‘communities’。
与 PhenoGraph 一样,我们首先基于 PCA 空间中的欧氏距离构建 KNN 图,并根据局部邻域中的共享重叠(杰卡德相似度)细化任意两个单元之间的边缘权重。此步骤使用 FindNeighbors() 函数执行,并将先前定义的数据集维度(前 10 个 PC)作为输入。
为了对cell进行聚类,我们接下来应用模块化技术,例如 Louvain 算法(默认)或 SLM,迭代地将细胞分组在一起,目标是优化标准模块化函数。 FindClusters() 函数实现此过程,并包含一个分辨率参数,用于设置下游聚类的“粒度”,增加的值会导致更多的聚类。我们发现,将此参数设置在 0.4-1.2 之间通常会为大约 3K 细胞的单细胞数据集带来良好的结果。对于较大的数据集,最佳分辨率通常会增加。可以使用 Idents() 函数找到簇。
pbmc <- FindNeighbors(pbmc, dims = 1:10)
pbmc <- FindClusters(pbmc, resolution = 0.5)
## Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
##
## Number of nodes: 2638
## Number of edges: 95965
##
## Running Louvain algorithm...
## Maximum modularity in 10 random starts: 0.8723
## Number of communities: 9
## Elapsed time: 0 seconds
# Look at cluster IDs of the first 5 cells
head(Idents(pbmc), 5)
## AAACATACAACCAC-1 AAACATTGAGCTAC-1 AAACATTGATCAGC-1 AAACCGTGCTTCCG-1
## 2 3 2 1
## AAACCGTGTATGCG-1
## 6
## Levels: 0 1 2 3 4 5 6 7 8
未完待续,持续更新,欢迎关注!
本文由 mdnice 多平台发布


)

DML)

)

用法详解)


)





)

)