更多资讯,请访问www.yinfotek.com 或关注微信公众号“殷赋科技”。殷赋学术交流2群已建立,需求加群的朋友,请在公众号输入“加群”,验证后即入群。
1
A:请教大家一个问题:在做对接模拟的过程中蛋白活性中心存在重要金属离子与配体之间的配位键相互作用,这个时候配位键应该如何处理?
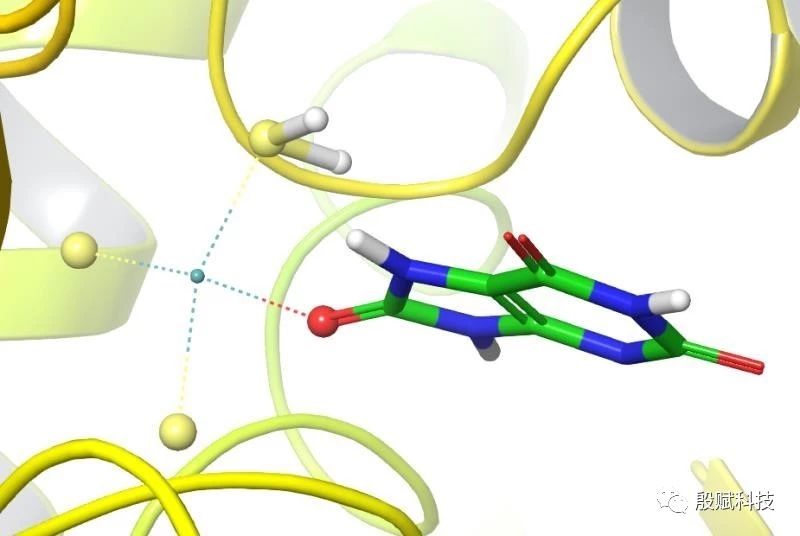
殷赋科技:你想怎样处理?
A:直接手动删除,然后添加构象限制?还是要采用共价对接的方式?
殷赋科技:不用管它,就留它在那。
C:是不是要重算分数电荷?
殷赋科技:分数电荷不好算,就让它保持正整数吧。
A:好的,一般情况下,这个金属配位键是不是要按照共价键来处理呢?
殷赋科技:为啥要这样处理呢,基于什么考虑?
D:共价键结合的是否无法做对接?
A:刚看到一篇文章,是这样描述的。

殷赋科技:不知道GOLD能不能做这种配位键的对接,一般按照非共价结合去对接就好。
D:金属配位这些,用计算机可能就搞不定了。
殷赋科技:如果以共价键来处理,那配体不就被你固定住了吗,那你不就知道配体的结合构象了吗,那还要对接干嘛,可是,你知道这个构象是对的吗、合适的吗?
金属与配体之间的相互作用,会在对接过程中考虑的,尽管软件可能不是以配位键来考虑,但以对接这种精度水平的方法来说,也差不多了。
殷赋科技:想要精确考虑,应该做动力学,甚至做QM/MM。
A:嗯,现在有很多做共价对接的方法和软件,比较AutoDock,好像是在形成共价键之前,先进行构象搜索和优化,然后再以正确朝向和位置的构象与氨基酸之间成键。是呢,QM/MM分层及计算。
殷赋科技:所以,你可以按照一般的非共价对接去做,找到正确的结合模式后,再手动将配位键连起来。
E: gold好像可以做带金属离子的对接。
A:Amber手册里也有比较详细的介绍。
F:amber我喜欢和terechem连用,可以用gpu算qm/mm。
G:还有ONIOM。
F:oniom不能算md。一般酶催化都用一些增强采样算法算pmf,这个能垒考虑了色散和熵,算的比用静态的过渡态能量准。
2
A:请问小分子和多糖的相互作用可以算吗?
殷赋科技:我司云计算平台可以计算。
A:通过动力学吗?
殷赋科技:分子对接即可。如果多糖简单,直接在平台上画出来,如果复杂,最好先找到晶体结构或者用其他软件方法模拟好构象。
A:只用分析对接会不会不够有说服力呀?我想问问有没有合适这种情况的分子动力学力场。
殷赋科技:根据你的研究目的选择方法,amber就有糖的力场。
3
A:群里有人会做基因富集吗?
B:我,你想怎么富集呢?
A:就是我预测了几靶点,然后知道对应的基因,想富集以后看主要是哪几个起作用大。
因为这个化合物活性不错,但就是不知道靶点。KEGG做过,但我不会分析。
C:用在线的KOBAS或者David,R语言的clusterProfile都行。
A:kobas我一直没有上上去,网页一直打不开,是不是我的网络有问题,还是什么原因?
D:如果是想看看而已 ,可以直接把这些基因扔string 就能出来go分析和kegg分析,很便捷。
A:string是?
D:https://string-db.org/cgi/input.pl?sessionId=4dMpPukDy9nJ&input_page_show_search=on。
C:做ppi的。
D:主要是做蛋白交互网络的,不过结果里也有go和kegg。
A: 做出来是这样。
D:
A:不用我设置什么吧,我就把我的基因丢进去,然后选择人源,然后continue。
D:是的。
A:这个主要是看哪几个关键信息?
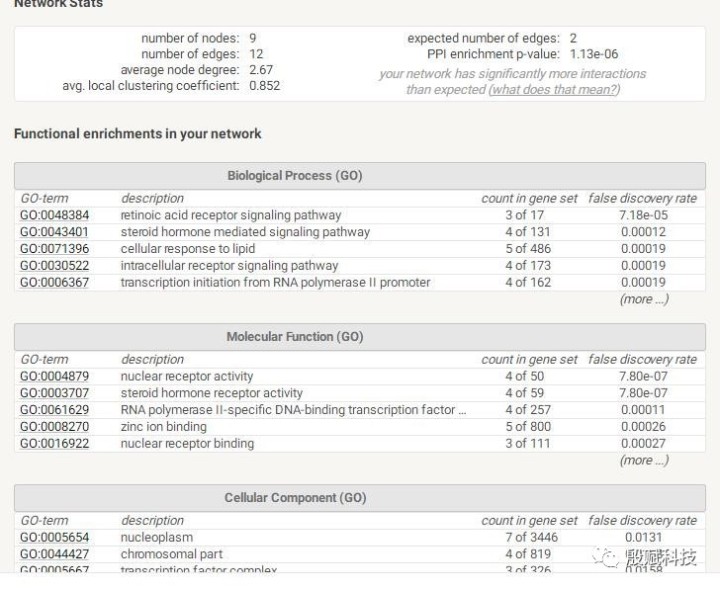
D:三个都看看 这是go的三种类别,能大约知道你这些基因主要是做什么的。
4
A:我也要请教个问题,结构相似性筛选,大家用什么软件做的啊?
B:碼代码。
A:除了代码呢?
B:不知道了。
C:请教下蛋白靶点预测有什么软件?
B:ds中的pp可以。
C:你不想代码,可以试下KNIME。
C:https://www.knime.com/knime-for-dBtB-scientists。
B:knime也需要自己搭流程。
A:ds的pp是啥呀?
B:和knime差不多的东西,但是里面有现成的可以用。
A:那有教程没?
C:你谷歌下。
A:我有DS软件,想用自己有的做。



![secure连不上远程地址_[笔记]Mariadb安装并配置远程访问](http://pic.xiahunao.cn/secure连不上远程地址_[笔记]Mariadb安装并配置远程访问)



)









-原创力文档...)


